

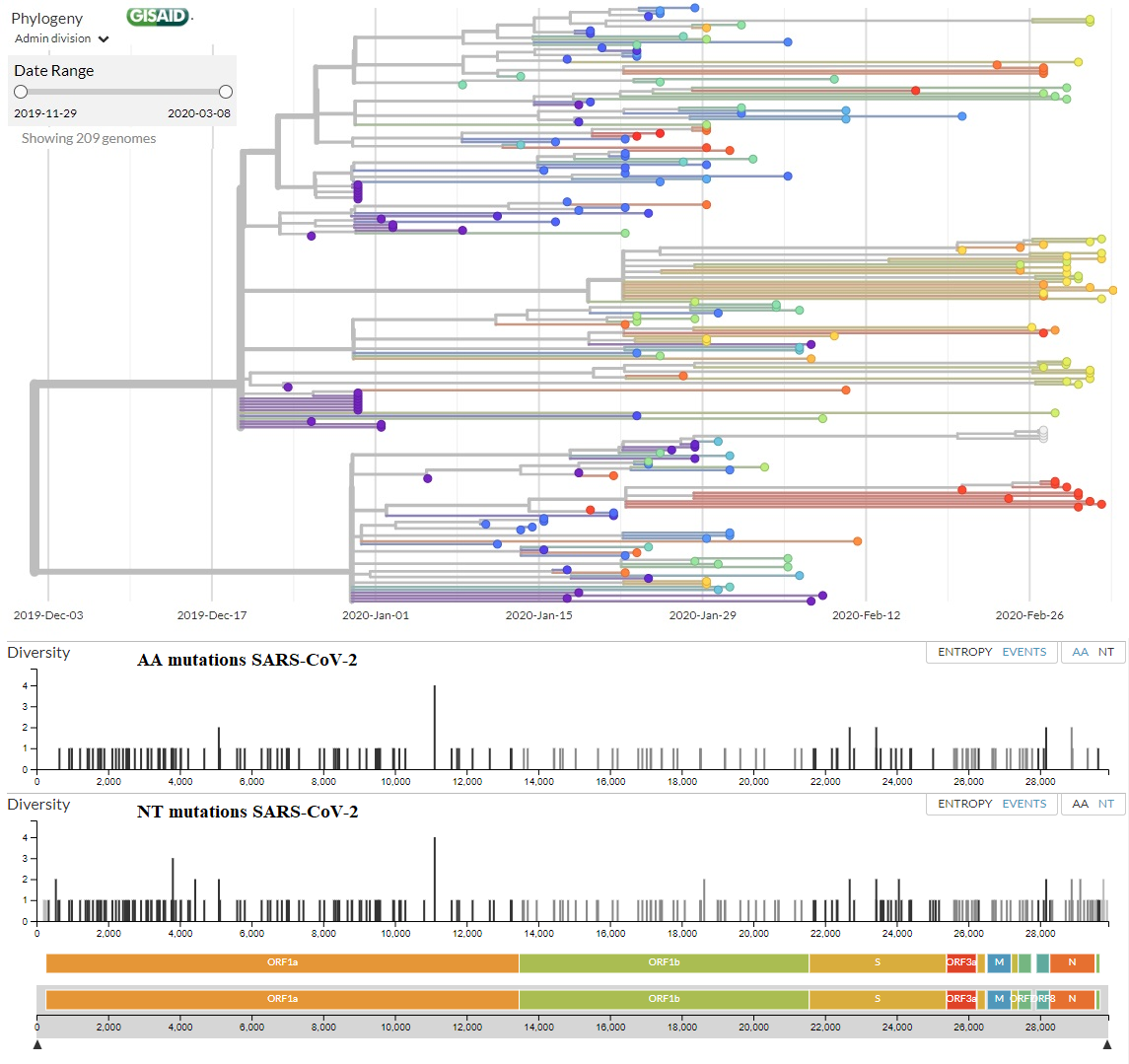
Quizás has leído por ahí que hay dos linajes (o cepas) del coronavirus SARS-CoV-2, llamadas L y S, que solo se diferencian en un único aminoácido mutado; S sería ancestro de L y ésta se transmitiría más fácilmente (sería prevalente en el ~70% de los casos). En realidad, lo único que hacen los virus de ARN es replicarse y mutar; por ello toda persona infectada con el coronavirus SARS-CoV-2 tendrá en su interior genomas víricos con diferentes mutaciones. Entre el 29 de noviembre de 2019 y el 7 de marzo de 2020 se han secuenciado 209 genomas completos de SARS-CoV-2, en los que se han observado 111 mutaciones de aminoácidos no sinónimas (recuerda que el código genético es redundante y las mutaciones ocurren en los nucleótidos, luego hay muchas más mutaciones de nucleótidos). Ninguna de estas mutaciones de aminoácidos tiene relevancia clínica demostrada en la infección por COVID-19; luego no se puede afirmar que haya dos cepas (o más) bien separadas. Así que no te dejes engañar, por ahora solo existe una cuasiespecie de coronavirus SARS-CoV-2 entre la población humana.
En esta figura te muestro el árbol filogenético (a fecha de 7 de marzo de 2020) con los 209 genomas secuenciados (la última versión actualizada la tienes en la web de NextStrain.Org, a partir de todos los genomas completos disponibles en la web de GISAID). En la parte inferior tienes todas las mutaciones de aminoácidos (AA mutations) y de nucleótidos (NT mutations) que se han encontrado, justo encima de los genes del coronavirus donde se han encontrado (puedes leer una explicación del genoma del SARS-CoV-2 en este blog en LCMF, 25 ene 2020). En el eje vertical tienes la diversidad de nucleótidos (Diversity) que mide el número promedio de diferencias entre nucleótidos en un sitio determinado entre dos secuencias ADN para todos los pares posibles en la población (más detalles y la fórmula que la calcula en la wikipedia). El listado de todas las mutaciones no sinónimas (de aminoácidos) lo tienes aquí, junto con el número de secuencias que la presentan.
Más información en Oscar A. MacLean, Richard Orton, …, David L. Robertson, «Response to “On the origin and continuing evolution of SARS-CoV-2”,» Virological.Org, 5 Mar 2020, quienes critican el artículo con la propuesta de las dos cepas de Xiaolu Tang, …, Jie Cui, Jian Lu, «On the origin and continuing evolution of SARS-CoV-2,» National Science Review, nwaa036 (03 Mar 2020), doi: https://doi.org/10.1093/nsr/nwaa036.

[PS 10 mar 2020] Como parece que en algunos institutos se está usando esta pieza para acompañar el tema de las mutaciones, creo que viene bien recordar que el código de colores del árbol filogenético corresponde a los países y regiones de origen, donde se ha extraído la muestra de virus secuenciada. Esta nueva figura se presenta el árbol para los 264 genomas del SARS-CoV-2 en GISAID hasta el 9 de marzo de 2020, así como el mapa con los países y regiones correspondientes. Espero que ayude a los docentes en su labor. Recomiendo la consulta directa de la web de NextStrain.Org para la última información disponible. [/PS]
[PS 17 mar 2020] Recomiendo leer a Mónica G. Salomone, «La genética traza el mapa de la dispersión mundial del virus. Los genetistas y biólogos computacionales tratan de dibujar el árbol genealógico de las mutaciones del SARS-CoV-2 a medida que se dispersa. El análisis de más de 500 genomas de los cinco continentes no muestra cambios en su virulencia por ahora, ni la aparición de linajes distintos», Agencia SINC, 16 mar 2020. [/PS]
[PS 26 ago 2020] Recomiendo leer a Takahiko Koyama, Daniel Platt, Laxmi Parida, «Variant analysis of SARS-CoV-2 genomes,» Bulletin of the World Health Organization 98: 495-504 (02 Jun 2020), doi: https://doi.org/10.2471/BLT.20.253591. Se analizan 10 022 genomas desde el 1 de febrero al 1 de mayo de 2020 de 68 países. Hay 5775 variantes respecto al genoma de referencia, 2969 mutaciones de aminoácidos, 1965 mutaciones sinónimas, 484 mutaciones en regiones no codificantes, 142 deleciones no codificantes, 100 mutaciones de marco de lectura, 66 inserciones no codificantes, 36 variantes que llevan a un codón de parada, 11 deleciones con cambio de marco de lectura y 2 inserciones con cambio de marco de lectura. Las mutaciones más comunes son la mutación sinónima 3037C >T (6334 muestras), la mutación P4715L en el marco abierto de lectura ORF1ab (6319 muestras) y D614G en la proteína espicular (6294 muestras). Se han identificado 6 clados (el basal, D614G, L84S, L3606F, D448del y G392D) y 14 subclados. La mutación entre bases más común es C >T con 1670 variantes distintas. [/PS]
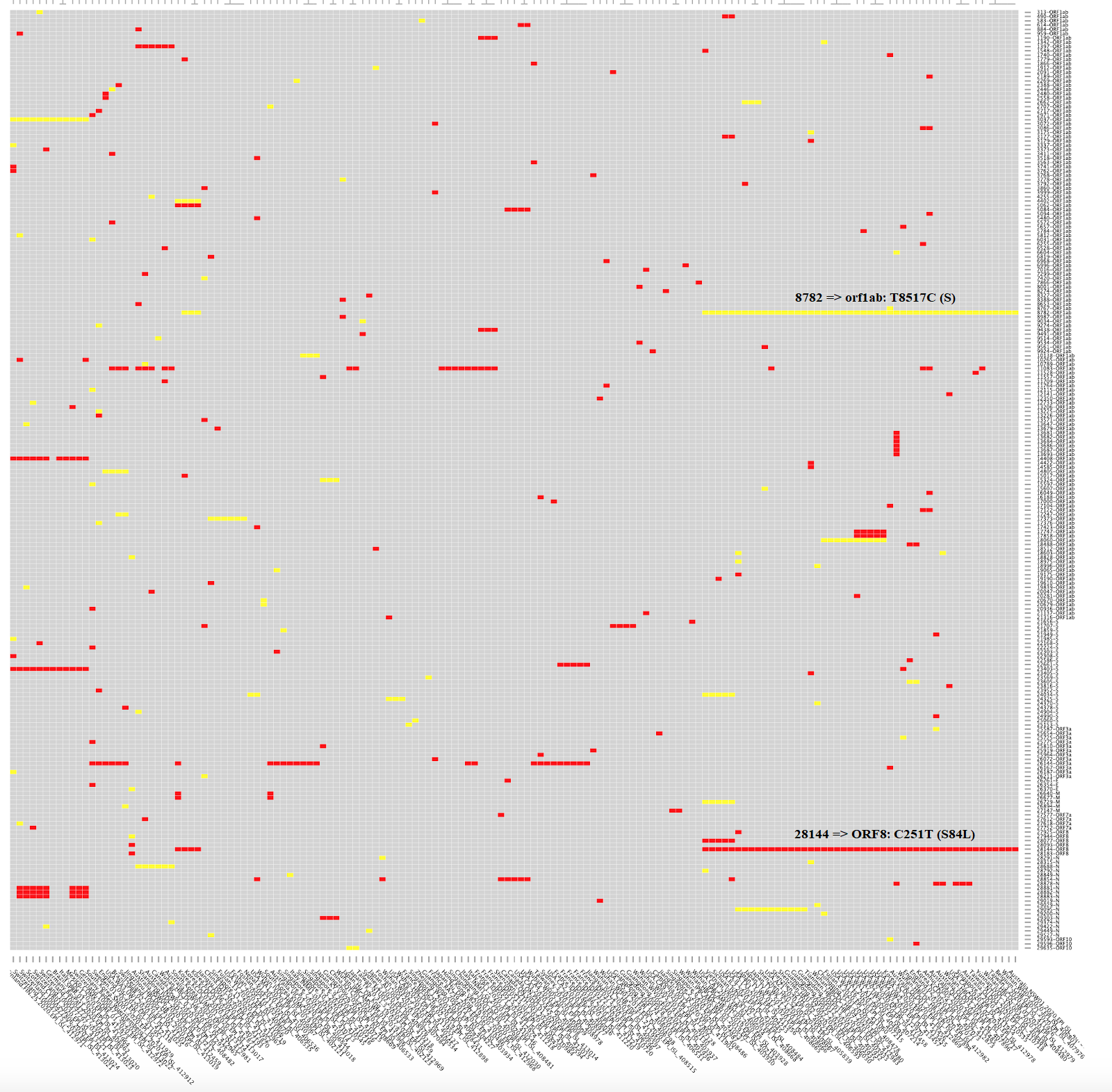
La crítica aún no publicada de MacLean et al. a la propuesta del artículo publicado por Tang et al. parece obvia para quien es consciente de que los coronavirus, como todos los virus de ARN, mutan mucho. Máxime cuando la propuesta es que la cepa S sea ancestro de la cepa L y que esta última se transmite más fácilmente (por ello, el ~70% de los infectados la tendría). Sin embargo, ambas cepas solo se diferenciarían en dos SNPs (se pronuncia esnips), polimorfismos de un solo nucleótido. En concreto, un SNP sinónimo en la posición 8782 del ARN, en el gen orf1ab, el cambio de una timina por una citosina en la posición 8517 de dicho gen (T8517C), que cambia el codón AGT de una serina por AGC también de una serina; y otro SNP no sinónimo en la posición 28144 del ARN, en el gen ORF8, el cambio de una citosina por una timina en la posición 251 de dicho gen (C251T), que cambia el codón de una serina por el de una leucina en la posición 84 (S84L). Esta figura muestra las 111 mutaciones no sinónimas en rojo y las restantes mutaciones sinónimas en amarillo; he aclarado dónde se encuentran las mutaciones 8782 (orf1ab: T8517C) y 28144 (ORF8: C251T, S84L).
La teoría evolutiva predice que el coronavirus SARS-CoV-2 está sometido a mutaciones continuas que producen una deriva genética que acabará conduciendo a su separación en diferentes cepas en el futuro. Lo habitual es que las cepas que mejor se adapten al ser humano (es decir, las que tengan una letalidad reducida y produzcan síntomas más leves) sean las que se acaben propagando con mayor facilidad. Pues al coronavirus le «interesa» sobrevivir el máximo tiempo posible entre los humanos y que no le pase lo que pasó con el SARS-CoV (cuya epidemia fue contenido y desapareció de la circulación entre los humanos).
Y, por cierto, seguro que en Twitter y en medios sensacionalistas has oído que el coronavirus SARS-CoV-2 podría haber escapado del laboratorio BSL-4 de Wuhan. Nada más lejos de la realidad. Se han publicado varios artículos que desmontan esta conspiración y demuestran que la variación genética natural de los betacoronavirus es suficiente para explicar la aparición de nuevos virus que puedan infectar a humanos (como ocurrió con SARS y MERS, y ahora con COVID-19). Lo que pasa es que las noticias virales como las del coronavirus acaban generando de manera espontánea gran número de conspiraciones, porque todos tenemos un poco de conspiranoicos aunque nos pese. Te recomiendo leer a Daniel Jolley, Pia Lamberty, El coronavirus es un campo abonado para los ‘conspiranoicos’,» The Conversation, 05 mar 2020.
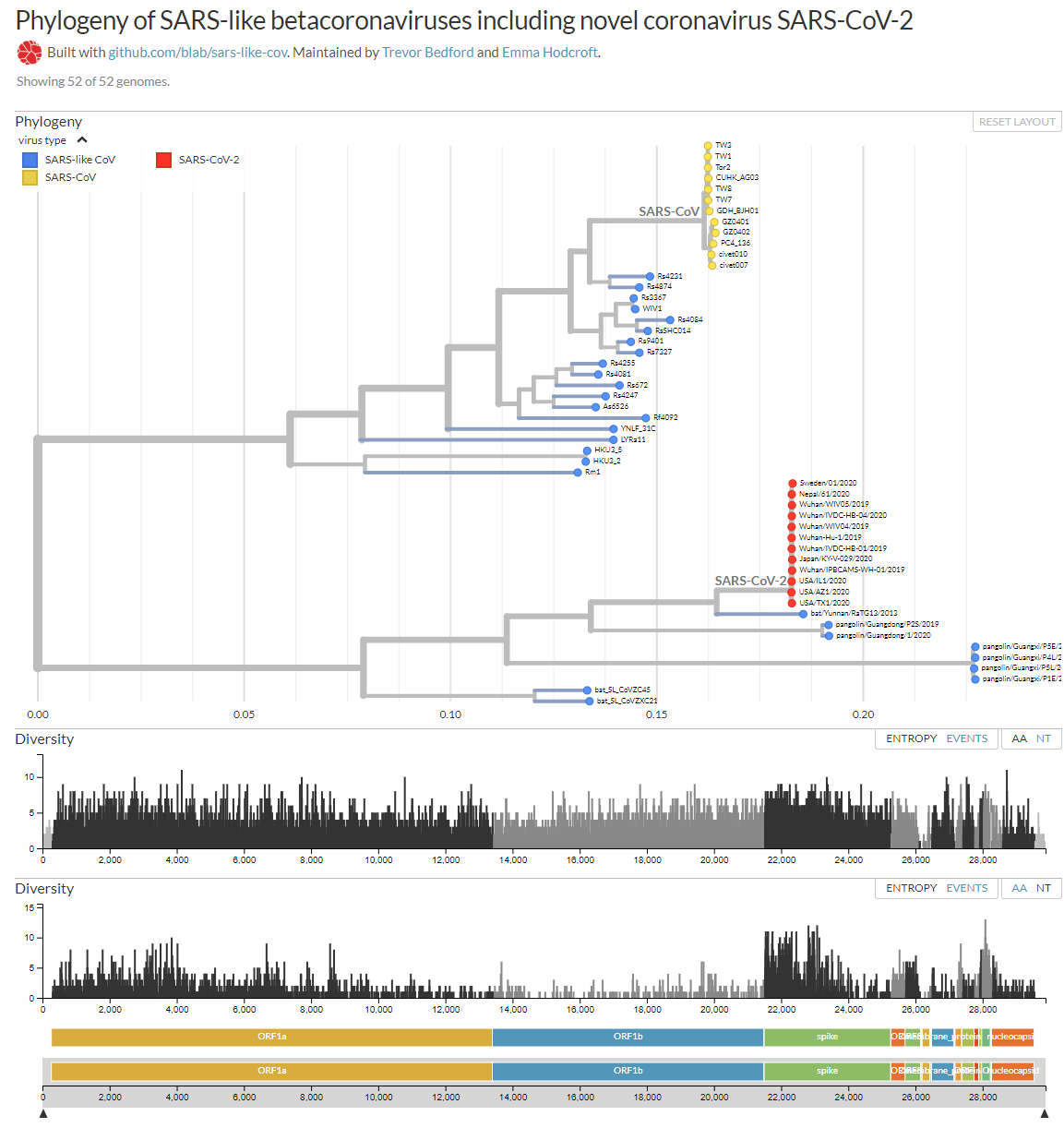
Me permito aportar mi granito de arena con esta figura que te muestra el arból filogenético de los betacoronavirus similares a SARS, entre ellos SARS-CoV-2, así como las mutaciones que se han observado en sus ARN. Prácticamente todos los nucleótidos (NT) han mutado alguna vez; y casi todos los aminoácidos (AA) también. Como se dispone de más genomas de los que infectan a humanos (SARS-CoV en amarillo y SARS-CoV-2 en rojo) que de los demás, se observa que la diversidad genética es mucho mayor en la glicoproteína S, responsable de la infección a humanos; pero su razón de ser es un simple sesgo estadístico. Como nos recuerda Ignacio López-Goñi en Twitter: «No, no se ha escapado de un laboratorio, ni es un arma biológica, la naturaleza se basta y se sobra para generar nuevos virus».
En resumen, el coronavirus SARS-CoV-2 está mutando de forma continua como cuasiespecie que es, pero todavía no hay datos suficientes para afirmar que haya más de una cepa. Que no te engañen ni te asusten con supuestas mutaciones que incrementan su letalidad; lo más habitual es que las mutaciones más letales, si algún día llegan a aparecer, no se propaguen entre la población. Lo peor que nos puede pasar es que el virus se acomode a los humanos y se haga estacional como el virus de la gripe; en dicho caso tendremos epidemias de COVID-19 todos los años. Por fortuna, habrá vacunas y en poco tiempo todo el mundo se olvidará de su existencia (como han hecho con el virus de la gripe A). Eso sí, mucha gente en grupos de riesgo no se vacunará (como no se vacunan de la gripe) y fallecerá (como fallecen por la gripe), pero sus decesos ya no serán noticia en los medios.
Hemos aprendido mas de este virus en solo 2 meses que otros que llevan milenios con nosotros, simplemente flipo al pensar lo que la ingeniería genética nos depara en la próxima década.
gracias Francis por el artículo.!
y totalmente de acuerdo con Alcalino: las cosas que se podría ver a medida que entendamos mas!
Hablando de las pruebas de evaluación para CNVs de densidad muy alta Roma, es a la qué algunos se refieren como. ¿vivaROMa, o Vivarma?
¿Que tan cierto es que exista una patente para este SARS-CoV-2?
Juan, se puede patentar la secuencia de ARN de un virus, aunque sea natural; parece un sinsentido, pero así lo permite la ley; el primero que secuencia el ARN lo puede patentar antes de publicarla; no me consta que SARS-CoV-2 (COVID-19) haya sido patentado (tuvo que enviarse la solicitud antes de la publicación y no me consta que haya sido enviada; si se envía después, será rechazada). Los coronavirus SARS-CoV (SARS) https://patentimages.storage.googleapis.com/6b/c3/21/a62eb55a0e678c/US7220852.pdf y MERS-CoV (MERS) https://data.epo.org/publication-server/pdf-document?pn=2898067&ki=B1&cc=EP&pd=20200115 fueron patentados.
No tengo ni idea sobre vivarma o vivaROMa.
Muchas gracias Francisco por tu respuesta.
Si el Co-vid 19 «se parece» al SARS Cov ….¿ podrán ser efectivas las vacunas ya desarrolladas para esos virus ?
Hernán, no hay vacunas contra el virus SARS-CoV, que desapareció de la circulación en humanos en 2003, así que nadie llegó a desarrollar nunca vacunas contra SARS. Se han desarrollado vacunas contra MERS-CoV, pero no sirven contra SARS-CoV-2. Aún así, la experiencia en el desarrollo de vacunas contra MERS ayudará a desarrollarlas contra COVID-19.
«Lo habitual es que las cepas que mejor se adapten al ser humano (es decir, las que tengan una letalidad reducida y produzcan síntomas más leves) sean las que se acaben propagando con mayor facilidad.» Uff menos mal, esperemos que no sea al contrario jeje.
Muy bueno. Me lo llevo a clase para Comentarlo
Con los alumnos de 4• ESO. Justo hemos visto mutaciones, vamos a empezar evolución y el tema no podía ser más actual y estar mejor explicado 👏🏼👏🏼👏🏼
Muy interesante y despeja mi duda sobre la capacidad de mutación de virus RNA y su adaptabilidad, una duda hay una parte de un link que sugieren consultar y aparecen varios países, entre estos Mexico(1), que significa el 1 entre ()
Significa que sólo hay un genoma secuenciado de un enfermo de Mexico (Mexico/CDMX-InDRE_01/2020).
Si el virus esta mutando continuamente como es q se explique q la vacuna sirva …si ya a mutado y tiene otro tipo de proteinas virales a lo largo del tiempo.
Y si se vuelve estacional ya no sera tan peligrosa?como lo son tantos tipos de gripe de invierno que las pasamos no tan facil pero con reposo y medicamentos ya sea naturales o de farmacia ,necesarios seguimos aqui vivitos.Para q entonces la vacuna??….otra vez para los mayores de 60 años y personas con enfermedades de base,q fueron las q se vacunaron años pasados y no les sirvio de nada pues fueron los primeros q cayeron enfermos y muchos muriron??un circulo vicioso sin sentido ,solo disminuyeron a la poblacion mas suceptible y debil con esas vacunas para la influenza.
Helper T, las vacunas se diseñan para exponer al sistema inmune regiones codificadas por el ARN del coronavirus que son muy estables y mutan mucho menos que el resto; además se usan métodos de desarrollo que permiten adaptar las vacunas a futuras mutaciones en dichas regiones (si llegan a aparecer); así se espera que las vacunas sean eficaces durante cierto tiempo (uno o varios años) y luego tengan que ser rediseñadas en sus detalles. Muchas farmacéuticas van a ofrecer las primeras vacunas a precio de coste, pero nadie espera que lo hagan con las futuros rediseños necesarios para lidiar con futuras mutaciones dentro de unos años.
Por otro lado, los virus que infectan a una especie por primera vez suelen evolucionar hacia cepas que producen menos síntomas y que tienen menor letalidad; pero no se pueda asegurar que vaya a ocurrir con el SARS-CoV-2 (al menos tras 9 meses no se observa que esté ocurriendo).
Hola, Francis. Quiero preguntarte si cabe la posibilidad de si los métodos de detección de este tipo de virus no puedan fallar. Salen muchos casos de Covid19 hasta en la sopa y hasta hace nada solo estaba en China. No podría confundirse con otros tipos de coronavirus?
Isalar, todos los experimentos tienen cierta tasa de fallos (falsos positivos) y los tests RT-PCR también (el porcentaje concreto depende de la variante del test que se use, pues hay varias). Aún así, hoy en día, la tasa de fallos es muy pequeña (menor del 1%).
Sería interedante nos compartieras en un post como funcionan los tests más usados de Covid_19. Así como saber cuáles coronavirus de humanos existen y hasta que punto la exposición con ellos pueden conferir inmunidad contra covid-19
Zovi, el tema de los tests lo dejo pendiente. En cuanto a la segunda pregunta se conocen 7 coronavirus que infectan a los humanos, 4 provocan resfriados (HCoV 229E, NL63, OC43, y HKU1), los otros 3 provocan SARS, MERS y COVID-19. Ninguno confiere inmunidad a ningún otro, salvo a sí mismos; así que el único que confiere inmunidad a COVID-19 es su causa, SARS-CoV-2.
https://twitter.com/emulenews/status/1239679714555551744?s=20
Estaria muy bien Francis un post sobre las secuelas que deja el Covid-19 en el cuerpo una vez curado, que hay mucha desinformacion al respecto.
Saludos y gracias!
Poselin, no siendo médico no soy la persona más adecuada para hablar de medicina; puedo hablar de genética y bioinformática, pero no me compete hablar de medicina clínica en este blog.
Espectacular mi consulta era con el clima ayuda a combatir este virus frio o calor
La vía de contagio mediada por el virus depositado en el ambiente se reduce con condiciones climáticas adversas para el coronavirus; sin embargo, la vía directa de contagio por proximidad se mantiene. Por ello el distanciamiento social y la autocurentena son fundamentales.
De un microbiologo molecular, MUCHAS GRACIAS por la compilacion e ilustrativa explicacion.. y mantener el rigor cientifico que a menudo se difumina en el sensacionalismo.
Saludos
Excelente Artículo, Francis.
Sin embargo, te ha faltado rematarlo con un titular bien vistoso.
Si le hubieses puesto «NO HAY EVIDENCIAS DE QUE HAYA VARIAS CEPAS DEL CORONAVIRUS» habrias logrado 10 veces mas visitas.
Es lo que tiene ser un científico honrado.
Francisco felicitaciones por este post realmente increible. Soy medico yquisiera hacer una consulta sobre la proteasa principal o sea la enzima clave para la replicación del coronavirus la Mpro, si es verdad que dicha enzima de tanto en SARS-CoV-2 como en SARS-CoV son altamente parecidas en términos de secuencias y estructuras. Pensando en un antiviral que se pueda utilizar. Eh leído sobre remdesivir o lopinavir y ritonavir (esto es un tema médico) pero estoy investigando sobre un antiviral más económico sólo que me estanque en la parte de la filogenética del virus. Desde ya te agradeceria tu respuesta. Gracias
Darío, no soy experto en estas lides. La secuencia de la proteína Mpro de SARS-CoV la tienes en https://www.ncbi.nlm.nih.gov/protein/29837498 y la de SARS-CoV-2 la tienes en https://www.ncbi.nlm.nih.gov/protein/1802476809 . La estructura 3D de Mpro de SARS-CoV se publicó en PNAS en 2003 https://www.pnas.org/content/100/23/13190.short y la estructura 3D para la Mpro de SARS-CoV-2 se ha publicado en https://www.biorxiv.org/content/10.1101/2020.02.26.964882v2 . Aparentan ser estructuras muy similares. Te copio aquí las diferencias entre las secuencias de aminoácidos de ambas.
SARS-CoV sgfrkmafps gkvegcmvqv tcgtttlngl wldd(t)vycpr hvict(a)edml
SARS-CoV-2 sgfrkmafps gkvegcmvqv tcgtttlngl wldd(v)vycpr hvict(s)edml
npnyedllir ksnh(s)flvqa gnvqlrvigh smqnc(l)l(r)lk vdt(s)npktpk ykfvriqpgq
npnyedllir ksnh(n)flvqa gnvqlrvigh smqnc(v)l(k)lk vdt(a)npktpk ykfvriqpgq
tfsvlacyng spsgvyqcam rpn(h)tikgsf lngscgsvgf nidydcvsfc ymhhmelptg
tfsvlacyng spsgvyqcam rpn(f)tikgsf lngscgsvgf nidydcvsfc ymhhmelptg
vhagtdleg(k) fygpfvdrqt aqaagtdtti t(l)nvlawlya avingdrwfl nrftttlndf
vhagtdleg(n) fygpfvdrqt aqaagtdtti t(v)nvlawlya avingdrwfl nrftttlndf
nlvamkynye pltqdhvdil gplsaqtgia vldmca(a)lke llqngmngrt ilgstilede
nlvamkynye pltqdhvdil gplsaqtgia vldmca(s)lke llqngmngrt ilgsallede
ftpfdvvrqc sgvtfq
ftpfdvvrqc sgvtfq
Quizás te interese leer (si no lo has hecho ya) «Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds,» Molecular Informatics (2020) https://doi.org/10.1002/minf.202000028 ; «Substrate specificity profiling of SARS-CoV-2 Mpro protease provides basis for anti-COVID-19 drug design,» bioRxiv (2020) https://doi.org/10.1101/2020.03.07.981928 .
Pero, repito, no soy experto en estas lides (solo imparto clases de bioinformática).
Espero que esta información te sirva.
Saludos, Francis
Muy buen aporte, llevo días investigando sobre el tema, y no había dado con esta web hasta ahora.
Hablando solo desde mi desconocimiento, ya que llevo solo 1 semana estudiando sobre el tema, entendemos no que son 2 mutaciones sinó como algunos autores explican que es una la evolución de la segunda?.
Respecto su orígen si parece que se han descartado varios animales, pero cobra fuerza la parte del murcielago, pero también he estado leyendo que es muy complicado que el virus pase del animal a la persona, es eso cierto?
También quería preguntar si al parecer la única solución está siendo medicamentos para el ébola ya que producen anticuerpos en grandes dosis, se estará usando esto para tratar a las personas contagiadas?
Por último, de la gente que se sabe que ha tenido el virus y no presenta ningún sintoma siendo «inmune» no se puede sintetizar algún tipo de vacuna de estas personas o utilizar esos anticuerpos de alguna manera?, a cuenta de todo esto he leído un artículo del año 2008 que hablan de un «atajo» en la creación de anticuerpos, haciendo en esencia que se puedan crear mas y en mejor cantidad. Dejo el artículo; https://www.rtve.es/noticias/20080430/descubierta-tecnica-rapida-para-generar-anticuerpos-monoclonales-completamente-humanos/41111.shtml ya que hablo desde mi mas completo desconocimiento y estoy aprendiendo a marchas forzadas en estos días.
Gracias si alguien me puede ayudar en lo que pregunto y repito, muy buen aporte
Hola Francisco, ya que estas en contacto con bioquímicos, sabes si se esta investigando sobre las personas de más de 99 años que se curaron de coronavirus en relación a la pandemia de gripe de 1918, ya que ellos o sus madres pudieron haberla pasado y generar algún tipo de defensa?
No me consta. Pero como debería ser obvio, la inmunidad a un virus de la gripe no ofrece inmunidad a un coronavirus. De hecho, ni siquiera ofrece inmunidad a otros virus de la gripe.
Muy interesante el artículo, aunque la verdad te perdiste la mejor parte que fué la discusión entre los científicos chinos que hicieron el estudio y los críticos que lo refutan, más abajo en los comentarios del link que compartiste. Los chinos aseguran que los críticos hicieron mal sus cálculos por lo que obtuvieron resultados erróneos y muestran otro estudio independiente que corrobora su información. Mientras que los críticos aseguran que el estudio de los chinos está sesgado desde la metodología y por la población pequeña que utilizaron. De cualquier manera, los chinos aceptan que el término «agresivo» es erróneo y lo sustituyen con «alta frecuencia» y demuestran que si hay secuencias sinónimas que se repiten aunque son pocas (que identificaron como «L» y «S» y las no sinónimas que son producto de la constante mutación del virus. Los críticos ya no respondieron al último comentario de los chinos acerca del error en sus cálculos por lo que deja con duda.
Hola, Muchas gracias por tus observaciones tan valiosas en este sentido.
Quisiera saber tu opinión sobre la noticia que nos comparten desde Corea del sur con 91 pacientes dados de alta que volvieron a enfermar (https://www.infobae.com/america/mundo/2020/04/10/alerta-en-corea-del-sur-el-coronavirus-se-reactivo-en-al-menos-51-pacientes-que-habia-sido-dados-de-alta/). Nos hablan de una posible «reactivación», además teniendo en cuenta este panorama, se ha demostrado en coronavirus felinos, que el fenómeno de ADE es posible.
Piensas que se posiblemente que los Ac no son neutralizantes y que además tenga un Rxn cruzada con alguna variante que ya tiene un cambio antigénico imprtante? o crees que se pueda deber a otra cuestión más aislada.
Gracias, para mi es importante este aprendizaje casi en «tiempo real» que como comunidad científica debemos hacer ante la crisis en salud publica.
Laura, hasta que no haya un artículo científico que discuta los detalles no puedo opinar. Las pruebas fallan y para explicar lo que ha pasado hay que conocer qué pruebas les hicieron para decidir que estaban curados (no puedo descartar que fallaran las pruebas). Supongo que no tardará en publicarse en medRxiv, pero hasta ahora lo único que encuentro son noticias de prensa que repiten una y otra vez lo mismo, todas dicen lo mismo, sin aclarar ningún dato relevante sobre el caso.
Recién veo el artículo.
Me surgen dos dudas a partir de lo que voy buscando e indagando sobre el genoma de SARS-CoV2..
Tenés idea qué % de similitud tiene el SARS-CoV2 (subespecie) con los otros coronavirus de la misma especie? dónde puedo buscar eso?
Tenés conocimiento sobre qué porción del genoma (tan mutante por cierto) están diseñados los cebadores/primers específicos para SARS-CoV2? Gracias!
Melisa, hay decenas de artículos con dicho porcentaje, p.ej. “The genetic sequence, origin, and diagnosis of SARS-CoV-2,” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7180649/, pero lo relevante es la comparación gen a gen, la sintenia (synteny) https://en.wikipedia.org/wiki/Synteny#Shared_synteny
¿Por qué a pesar de existir múltiples mutaciones, en la mayoría de los casos el resultado fenotípico del virus SARS-COV-2 será el mismo?. Qué tipo de mutación se esperaría para que cambie algún producto del virus?
Hola Francisco me puedes ayudar con esta pregunta por favor?
¿Por qué a pesar de existir múltiples mutaciones, en la mayoría de los casos el resultado fenotípico del virus SARS-COV-2 será el mismo?. Qué tipo de mutación se esperaría para que cambie algún producto del virus?
Luciana, las mutaciones de los virus que son relevantes desde un punto de vista clínico son las que introducen nuevas vías de infección, las que evaden la acción de los fármacos que usan como tratamientos y las que minimizan los síntomas logrando que se incremente el número de casos asintomáticos. A pesar de que cinco meses nos perece un tiempo infinito, y que se han publicado más 18 000 artículos sobre la COVID-19, cinco meses es un tiempo insuficiente para saber qué mutaciones son relevantes desde un punto de vista clínico. A finales de este año, con la segundo rebrote, seguramente aprenderemos mucho sobre estas mutaciones.
No sé si conoces el caso de la estadística en la II Guerra Mundial. Ahora mismo las partes más relevantes del genoma del coronavirus no están mutando (o mejor dicho, dichas mutaciones no se propagan), o están mutando muy poco. SARS-CoV-2 muta menos que otros coronavirus, porque tiene una proteína (nsp14-ExoN) que actúa como una enzima capaz de reparar los errores que pueden ocurrir durante la replicación del genoma; por ello, de momento, no está acumulando mutaciones que afecten a su virulencia.
Felicidades, la información las ligas, no ayudan a comprender mejor el complejo mundo de los coronavirus, no cabe duda ellos y nosotros seguiremos en el camino de la evolución