

Todo apunta a que el sarbecovirus SARS-CoV-2 (responsable de la COVID-19) divergió del reservorio natural de sarbecovirus de murciélagos hace varias décadas. Se publican en Nature Microbiology tres estimaciones de máxima verosimilitud para su divergencia respecto a Bat-CoV-RaTG13, basadas en usar tres regiones no recombinantes diferentes: la fecha más conservadora es 1969 (intervalo 1930–2000 al 95 %) para NRR1, pero no hay que descartar 1982 (1948–2009 al 95 %) para NNR2, y 1948 (intervalo 1879–1999 al 95 %) para NRA3. Para la divergencia respecto al coronavirus de pangolín se estima el rango entre 1851 (1730–1958) y 1877 (1746–1986); parece que otro animal tuvo que ser el intermediario entre murciélagos y humanos, si lo hubo. Más aún, el ancestro común de los sarbecovirus SARS-CoV (responsable del SARS) y SARS-CoV-2 divergió hace muchos siglos; se estima que una fecha alrededor del año 1200 ± 400. Toda una sorpresa para quienes creían que esta divergencia era muy reciente.
Por supuesto, hay que tomar con mucho cuidado estas estimaciones. Los coronavirus son muy recombinogénicos; cuando dos diferentes infectan la misma célula hay trozos de ARN de algunos de sus genes homólogos que se recombinan durante la replicación. Así se explica que el dominio de unión al receptor (RBD) de la proteína espicular (S) del SARS-CoV-2 se parezca más al RBD de un coronavirus de pangolín que al del coronavirus de murciélago (RaTG13) al que más se parece el resto de su ARN. Cuando se usa la hipótesis del «reloj molecular» para estimar la fecha de las divergencias en árboles filogenéticos hay que eliminar las regiones que se han recombinado, pues su presencia contradice la hipótesis de que las mutaciones en proteínas homólogas (que tienen un ancestro común) ocurren a un ritmo constante a lo largo del tiempo.
En el nuevo artículo se han identificado tres regiones en los 68 genomas de sarbecovirus estudiados que parecen estar libres de recombinaciones (NNR1, NNR2 y NRA3); así se han obtenido sendas estimaciones de las fechas de divergencia. Solo con este estudio es imposible saber cuál es la más fiable; aún así, como todos los intervalos de fechas son coherentes entre sí, se puede inferior con rigor que la divergencia ocurrió hace entre 40 y 70 años. El artículo es Maciej F. Boni, Philippe Lemey, …, David L. Robertson, «Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic,» Nature Microbiology (28 Jul 2020), doi: https://doi.org/10.1038/s41564-020-0771-4, bioRxiv preprint 015008 (31 Mar 2020), doi: https://doi.org/10.1101/2020.03.30.015008.
En español recomiendo leer a Mónica G. Salomone, «El SARS-CoV-2 lleva entre 70 y 40 años en los murciélagos y lo más probable es que saltara directamente a humanos», Agencia SINC, 30 jul 2020.

La filogenia molecular es una técnica que tiene mucho más de arte que de técnica. Los árboles filogenéticos se construyen a partir de proteínas homólogas (que tienen un ancestro común), así que el resultado depende mucho de cuáles se consideren más representativas de los organismos considerados. Además, tanto para el alineamiento múltiple de las secuencias de dichas proteínas (o trozos de proteínas), como para la reconstrucción del árbol filogenético se pueden usar varios algoritmos diferentes; ninguno es mejor que otro, aunque los mejores «artistas» suelen obtener resultados coherentes.
Los coronavirus tienen unas treinta y pico proteínas (por ejemplo, SARS-CoV-2 tiene 36 en la anotación en GenBank de su genoma de referencia, de las que 26 son péptidos que están en la poliproteína orf1ab). Así, dependiendo de las proteínas (o trozos de proteínas) que se usen se obtienen resultados muy diferentes. Esta figura muestra cinco árboles filogenéticos inferidos a partir de diferentes regiones del alineamiento múltiple de los 68 genomas de coronavirus estudiados en este nuevo artículo. Se observa claramente que hay grandes diferencias entre ellos. La causa es la existencia de recombinaciones entre sus genomas. Así que hay que aplicar métodos de eliminación de segmentos genómicos recombinantes; también hay varios métodos diferentes para hacerlo.
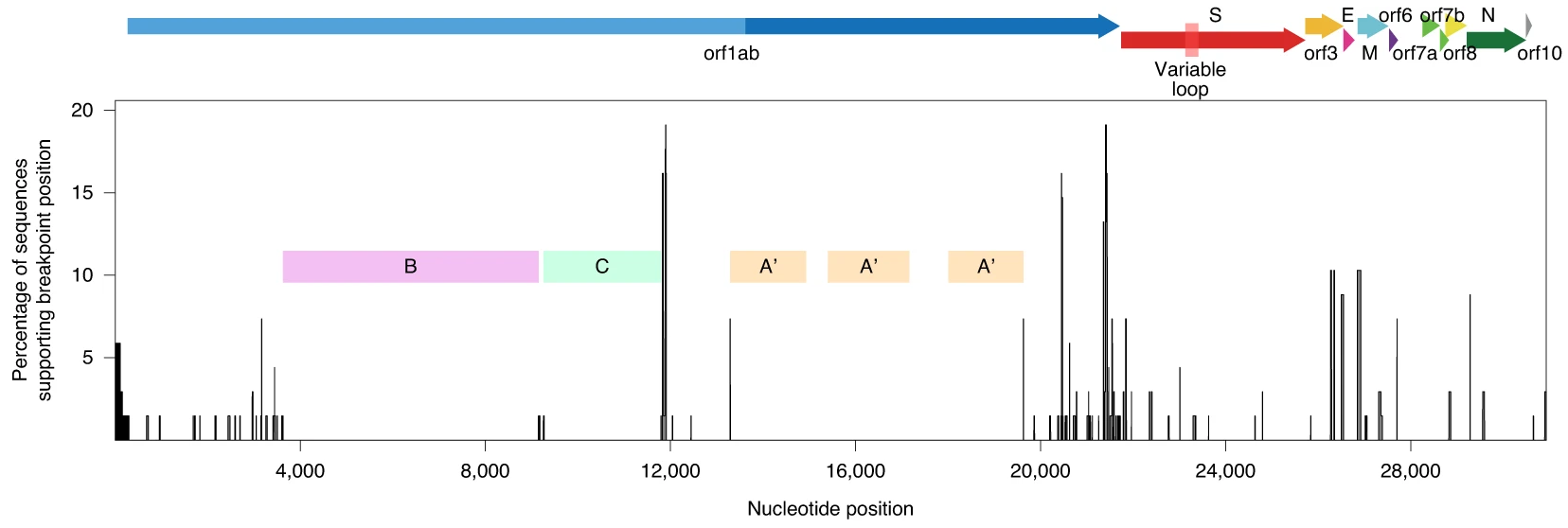
Por tanto, el resultado de usar el «reloj molecular» en el análisis de árboles filogenéticos de coronavirus depende mucho de las regiones libres de recombinaciones (NRR) que se seleccionen. Las barras verticales en esta figura son los puntos de ruptura (breakpoints) donde se estima que han ocurrido recombinaciones. Como puedes observar, la mayoría de las proteínas de estos sarbecovirus aparentan haber sufridos eventos de recombinación. En el nuevo artículo se han identificado cinco regiones que parecen libres de recombinaciones (NRR) pues están libres de puntos de ruptura: las tres regiones A’ (nt 13291–14932, 15405–17162 y 18009–19628), la región B (nt 3625–9150) y la región C (9261–11795); la región concatenada A’BC se ha llamado NRR1 y se considera la más conservadora. También se han considerado otras dos regiones libres de recombinación: NNR2 (nt 11885–21753) identificada con el método 3SEQ y NRA3 (el artículo no aclara cuáles son sus nucleótidos supongo que porque están repartidos por todo el genoma) identificada con el algoritmo RDP5. Los interesados en más detalles técnicos deberían consultar el artículo.
En resumen, se ha estimado la fecha de la divergencia entre el nuevo coronavirus humano y el coronavirus de murciélago más próximo; ocurrió hace entre 40 y 70 años. Además se ha estimado la fecha de divergencia con el coronavirus de pangolín más próximo y con el otro sarbecovirus humano. Las fechas de estas estimaciones deben ser tomadas como aproximadas; este tipo de análisis filogénticos basados en el reloj molecular tienen mucha incertidumbre. Conforme se publiquen nuevos análisis con un mayor número de secuencias de coronavirus se podrá identificar mejor las regiones que presentan recombinaciones y cuáles están libres de ellas, con lo que se podrán obtener estimaciones más confiables.
Como cientófilo, una de las cosas que me mueve últimamente a informarme sobre coronavirus, es tener material para dar estopa a la sarta de conspiranoicos e iluminados salvamundos que está saliendo por todos lados en un intento por entorpecer el avance del conocimiento científico, especialmente en este virus, que tanto nos está dañando. Como doctor en Biología alucino y me alegro enormemente precisamente por el hecho de que este avance esté siendo espectacular. Visto en perspectiva, hablamos de meses para generar un artículo como este, cuando hace 15 años se hubiera tardado toda una tesis. ¡Bien por la ciencia!!
Al hilo del comentario anterior, que se cogratula de tener un potente arsenal anti-conpiranoico, he analizado 57 de los coronavirus del artículo citado, todos ellos procedentes de murciélagos habitando su medio natural en diversas regiones de China. En concreto su proteína S y dentro de ella el motivo canónico de unión a integrinas, el tripéptido RGD. No lo presenta ninguno. Lo más próximo a ese motivo, y que también tiene alguna capacidad, bien qué débil o enfocada en una integrina muy específica, es el KGD, presente en un 19.3% de las muestras. En el resto de estos coronavirus, en la misma posición aparecen RSS (52.6%) y RFS (28.1%). ¿Y qué hay de Bat RaTG13?¿Dónde ubicarlo?¿Qué tripéptido porta en esa posición? Este virus es muy especial. Descubierto en 2013 no recibe su puesta de largo hasta enero de 2020, con la epidemia ya en desarrollo. Luego se indica que practicamente constituye el «backbone» de Sars CoV 2. Pues bien, en el lugar señalado no lleva ninguno de los motivos qué portan sus homólogos víricos rescatados de la naturaleza durante casi dos décadas. Ahí lleva TGD. Una secuencia inédita en el medio silvestre. No he detectado ningún betacoronavirus natural que presente ese mismo tripéptido. Y si no lo hay en la naturaleza, ¿De donde puede provenir? Aquí dejo la pregunta para qué alguien más competente que yo me la responda. Gracias.