

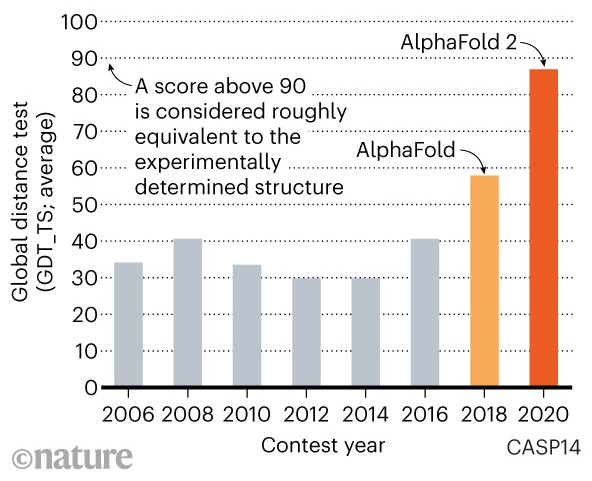
La estructura determina la función de una proteína; así, el problema del plegamiento de proteínas es clave para determinar su función biológica. Cada dos años, desde 1994, las últimas versiones de los software más avanzados compiten entre sí en el concurso de predicción del plegamiento de proteínas CASP (Critical Assessment of protein Structure Prediction). AlphaFold de Deepmind (Google) obtuvo el primer puesto en el CASP13 de 2018 con una puntuación de 120.4, por encima de 107.6 y 99.4 para el segundo y tercer puestos, resp. Este año, AlphaFold 2 ha vencido y ha arrasado en el CASP14 con una puntuación de 244.0, por encima de 90.8 y 89.0 para el segundo y tercer puestos. El aprendizaje profundo con redes de neuronas artificiales está revolucionando este área; de hecho, tras la victoria de AlphaFold en CASP13, la mayoría de los software concursantes en CASP14 usan diferentes versiones de dicha técnica. Sabemos muy poco de los detalles, ya que todavía no se ha publicado el resultado en un artículo científico (se rumorea que está en revisión para la revista Nature).
Para casi dos tercios de las proteínas las predicciones de AlphaFold son indistinguibles de las observaciones con cristalografía en rayos X o con criomicroscopia electrónica, los dos gold standard experimentales; incluso podría ocurrir que la predicción fuese más precisa que la observación experimental en algunos casos. La competición dura varios meses durante los que se van proponiendo unas cien objetivos (proteínas completas o dominios de proteínas), dando un plazo de varias semanas para que los competidores ofrezcan su predicción; una vez se supera el tiempo límite, un grupo independiente de científicos evalúa la proximidad entre cada predicción (anonimizada para no sesgar su juicio) y la estructura determinada de forma experimental. De hecho este año la mayoría de los software han realizado predicciones muy buenas; AlphaFold en muchos casos ha superado 90 de 100 puntos, mientras otros software se han quedado alrededor de 75 puntos. Por cierto, casi el 50 % de los equipos usa aprendizaje profundo (deep learning).
Por cierto, a principios del año 2020, AlphaFold realizó predicciones para algunas de las proteínas del nuevo coronavirus SARS-CoV-2; para una de ellas, ORF3a, que tiene 275 aminoácidos, su predicción acabó siendo casi idéntica a la determinada por criomicroscopia electrónica; en el CASP14 se usó otra proteína del coronavirus, ORF8, con 121 aminoácidos, para la que también se obtuvo una buena predicción. Más información en Ewen Callaway, «‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Google’s deep-learning program for determining the 3D shapes of proteins stands to transform biology, say scientists,» News, Nature (30 Nov 2020), doi: https://doi.org/10.1038/d41586-020-03348-4; The AlphaFold team, «AlphaFold: a solution to a 50-year-old grand challenge in biology,» DeepMind Blog, 30 Nov 2020; Robert F. Service, «‘The game has changed.’ AI triumphs at protein folding,» Science 370: 1144-1145 (04 Dec 2020), doi: https://doi.org/10.1126/science.370.6521.1144.
El funcionamiento de AlphaFold y sus resultados en CASP13 se publicaron en Andrew W. Senior, Richard Evans, …, Demis Hassabis, «Improved protein structure prediction using potentials from deep learning,» Nature 577: 706-710 (15 Jan 2020), doi: https://doi.org/10.1038/s41586-019-1923-7; más información divulgativa en Mohammed AlQuraishi, «A watershed moment for protein structure prediction,» Nature 577: 627-628 (15 Jan 2020), doi: https://doi.org/10.1038/d41586-019-03951-0; Arunima Singh, «Deep learning 3D structures,» Nature Methods 17: 249 (04 Mar 2020), doi: https://doi.org/10.1038/s41592-020-0779-y. El código fuente en Python 3.6+, usando TensorFlow 1.14 (que no es compatible con TensorFlow 2.0+) se encuentra en GitHub deepmind-research/alphafold_casp13/.
[PS 08 dec 2020] Recomiendo de forma encarecida la lectura de Carlos Outeiral Rubiera, «CASP14: what Google DeepMind’s AlphaFold 2 really achieved, and what it means for protein folding, biology and bioinformatics,» Oxford Protein Informatics Group (OPIG), 03 Dec 2020. Destaca que, en en la práctica, AlfphaFold 2 trabajó con recursos ilimitados de Google; se estima que consumió dos órdenes de magnitud más recursos computacionales que sus competidores, con un coste estimado en muchos millones de dólares. Además, AlphaFold 2 se entrenó con proteínas del PDB que tienen un sesgo hacia las proteínas fáciles de cristalizar; por tanto, para proteínas difíciles de cristalizar sus resultados puede que no sean tan buenos. Finalmente, no ayuda a resolver el gran problema más allá de la predicción de la estructura, determinar las interacciones proteína-ligando y proteína-proteína. [/PS]

La puntuación media de AlphaFold 2 en la competición CASP14 ha sido de 87.0 puntos sobre 100, con una mediana de 92.4 sobre 100; se estima que el error cuadrático medio de sus predicciones es de 1.6 Å (ángstrom), valor comparable con la anchura media de un átomo (1 Å, o sea, 0.1 nm). Entre las proteínas de la categoría de mayor dificultad, AlphaFold 2 logra una mediana de 87.0 puntos. Unos resultados espectaculares, sin lugar a dudas, que han sorprendido a muchos expertos.

Esta figura resume la arquitectura de la red neuronal de aprendizaje profundo usada en AlphaFold 2, que ha sido entrenada con todas las estructuras de proteínas publicadas en el PDB (Protein Data Bank), ~170 000 estructuras de proteínas, junto con otras bases de datos de secuencias de proteínas de estructura desconocida. Se usan dos redes de neuronas artificiles conectadas entre sí; una, en rojo en la figura, recibe como entrada el resultado del alineamiento múltiple de la secuencia (MSA) a partir de las bases de datos disponibles (usando una herramienta similar a BLAST) y la otra, en verde en la figura, recibe directamente la secuencia de aminoácidos; ambas tienen múltiples capas (en la figura solo se muestran las dos primeras). Ambas redes están realimentadas y su resultado combinado corresponde a la estructura de la proteína, junto con una puntuación asociada a la confianza estadística esperada y una matriz de distancias entre pares de aminoácidos de la secuencia.

AlphaFold 2 se ha implementado en 16 TPU v3s, cada una con 8 núcleos (cores) y una memoria de 128 GiB; esto equivale a entre ~100–200 GPUs. Recuerda que las TPU (Google’s Cloud TPU) son unidades de procesado tensorial para ejecutar algoritmos de aprendizaje profundo mediante TensorFlow). Durante la competición se ha usado un tiempo total de varias semanas. Los detalles de las capas, número de neuronas y demás detalles se publicarán en un futuro artículo (que según los rumores en Twitter ya está en revisión para la revista Nature).
En resumen, en cuatro años AlphaFold ha pasado de una idea de concepto a ser el software más potente para la reconstrucción de estructuras de proteínas. Sin lugar a dudas, el futuro del aprendizaje profundo en la reconstrucción de la estructura tridimensional de proteínas es muy prometedor. Habrá que estar al tanto de las futuras publicaciones sobre el tema y de los avances en las futuras competiciones CASP. Son muchas las aplicaciones potenciales en biomedicina y el potencial retorno económico para una empresa especializada en este área es grande. Solo el futuro dirá hasta dónde podemos llegar con las futuras versiones de AlphaFold de DeepMind.
Hola Francis muy buen articulo
Tengo varias preguntas
1. esto nos ayuda a entender como funciona el plegamiento de proteinas? o sea de terciaria o etapa cuaternaria? o no nos dice nada ?
2. solo con la estructura 3d ya tenemos la funcion de una proteina? no entiendo por que entonces salen todo el tiempo articulos explicando funciones de proteinas que ya conocemos su estructura 3d y hasta otrs funciones?
3. un futuro alpha fold 3 etc eliminara la necesidad de usar cristalografia Rx o criomicroscopia electronica? o aun la necesidad de simuladores cuanticos para ver como interactuan proteinas?
gracias
(1) no, por desgracia no ofrece ninguna información al respecto; (2) la función depende de los sitios activos de la proteína y de sus interacciones con metabolitos en dichos sitios activos; aunque la estructura determina la función, determinar la función a partir de la estructura es un problema diferente para el que AlphaFold no aporta nada; (3) no lo creo, en mi opinión convivirán dichas técnicas con pocos conflictos; la razón es que hay proteínas muy inestables que son casi imposibles de observar con cristalografía o con criomicroscopia, para ellas la predicción teórica es la única opción disponible.
donde puedo leer como funciona exactamente estas dos redes?
Aca analizan el paper anterior
https://www.youtube.com/watch?v=B9PL__gVxLI&t=2575s&ab_channel=YannicKilcher
Cuando se publique el paper podrás leerlo, hasta entonces nadie conoce los detalles. Mientras tanto, cualquier manual de TensorFlow te dará información suficiente para ir haciendo tus pinitos.
Creo que siguiendo el método científico, una predicción siempre tendrá que ser comprobada, por tanto dudo que de momento las predicciones puedan sustituir a la cristalografía, creo que pueden ser buenas aproximaciones, pero tendrán que ser comprobadas.
Podríais poner algunos ejemplos de las aplicaciones de estas investigaciones?
Fran, AlphaFold 2 todavía no ha sido usado en aplicaciones prácticas (ni siquiera se ha publicado un artículo científico que discuta el prototipo). Siendo trivial pedir «algunos ejemplos de aplicaciones» de un prototipo, nunca se debe olvidar lo que significa un prototipo.
Me pregunto si estos éxitos de la IA no nos irán poco a poco alejando de comprender las cosas.
Me recuerda a lo que ha pasado en ingeniería con los programas de elementos finitos.
Para la mayoría, acabas utilizándolos para todo y olvidas hasta cómo calcular un equilibrio de fuerzas y momentos.
Sin embargo, bien utilizados, ayudan mucho a comprender cómo trabaja una estructura y a decidir cuál es la mejor idealización para entenderla.
Cierto, amago, pero creo que esto es distinto, porque hoy por hoy (corregidme por favor si estoy desactualizado) no se comprende por qué las proteinas «eligen» una u otra forma. Y en el momento en que se pierde el interés en buscar unas reglas heurísticas, se pierde un montón de fuerza de conocimiento.
En otros campos ocurre lo mismo; hoy por hoy nadie va a perder el tiempo, por ejemplo, buscando una física capaz de predecir la mejor forma de enfriar rápidamente una botella o lata de líquido con una forma geométrica arbitraria (si tal cosa existe), hoy todo el mundo lucha por conseguir la mejor red neuronal para ello y conseguir el preciado «frigoondas»…y se conseguirá, y nunca se sabrá si tal física existe. Y es el primer ejemplo que me viene ahora, el cual no es muy relevante, pero en astrofísica o biología hay más ejemplos.
Tal vez, en el futuro, deje de tener importancia el «comprender», quien sabe, tal vez entre las demostraciones a base de fuerza bruta, los ordenadores cuanticos oráculos y la IA, lleguemos a la conclusión de que lo que nosotros llamábamos comprender, pues tampoco era gran cosa, y que una red neuronal es tan buena como un montón de ecuaciones…quien sabe..
La siguiente etapa sería, de la función requerida pasar a la estructura y de la estructura a la cadena de aminoácidos, sería la revolución de los biomateriales.
Ya existen ejemplos de diseño exitoso de proteínas. https://www.nature.com/articles/d41586-019-02251-x
Wow muy interesante, pregunta sr.Francisco R. Villatoro tienes una biografia de usted me interesaria saber como se convirtion en fisico e ingeniero. que buen blog.
Lo resumo aquí https://naukas.com/2020/11/23/francis-villatoro-en-espana-faltan-referentes-de-la-ciencia-que-tambien-lo-sean-para-la-sociedad/
Algun dia conocere a Francis es mi rockstar!! saludos desde Perú
Ok entiendo eso de la prediccion de proteinas, mejor comprendemos su funcion y creacion, mejor podemos crear curas o soluciones a enfermedades en medio a largo plazo.
ahora el hardware: Corre Crisis? (chiste gamers)
Excelente noticia, sin lugar a dudas. Sólo un pequeño comentario crítico con respecto a estas técnicas, aunque hablo sin tener mucha idea porque mi formación es más bioquímica-biológica que biofísica, y ya sabemos que el tema del plegamiento de proteínas es más física que otra cosa.
Entiendo que AlphaFold2 ha usado datos ya existentes de estructuras determinadas experimentalmente, sea con CryoEM, NMR o cristalografía, y la red neuronal «aprende» a partir de ejemplos, por tanto en mi opinión no sería adecuado para predecir de forma exacta el plegamiento a partir de una determinada cadena de aminoácidos. Es una grandísima noticia, y espero que pongan este software a disposición de la comunidad pronto.
Mi pregunta, para alguien más entendido, sería posible hacer una simulación más precisa de todo el proceso de plegamiento, y no sólo la estructura final, haciendo simulaciones en ordenadores cuánticos, que trabajan mejor con estos sistemas? para mí eso sería la panacea, y además supongo que se podrían implementar algoritmos de docking para evaluar mejor las interacciones proteína-proteína y proteína-ligando. Vamos, que veo esto como un parche, un parche excelente y muy necesario que nos ahorrará muchas horas de experimentación, pero siempre se puede mejorar.
Plegador novato, los ordenadores cuánticos son el futuro del docking de fármacos (interacción ligando-proteína y proteína-proteína), aunque su rol en el plegamiento de proteínas no está tan claro (dado la complejidad del entorno en el que se traduce la proteína y se pliega durante la traducción, la decoherencia cuántica tiene una escala de tiempo muy corta, mucho más corta que la escala de tiempo del plegamiento, lo que permite augurar que una aproximación cuasiclásica debería ser suficiente, así que bastaría química cuántica en ordenadores clásicos para resolver los detalles del proceso de plegamiento, sin necesidad de física cuántica en ordenadores cuánticos). En cuanto a la comprensibilidad (understability) en aprendizaje profundo, se está trabajando mucho en este tema; quizás en una década se rechace toda red de neuronas artificiales que no explique cómo obtiene sus resultados usando una explicación comprensible para los expertos en el área y para otras redes de neuronas artificiales; el uso de algoritmos de tipo «caja negra» (black box) conlleva una enorme pérdida de robustez en entornos reales.
Recomiendo la lectura del artículo de revisión de Carlos Outeiral, Martin Strahm, …, Charlotte M. Deane, «The prospects of quantum computing in computational molecular biology,» WIREs Comput. Mol. Sci. 11: e1481 (2021), doi: https://doi.org/10.1002/wcms.1481, arXiv:2005.12792 [quant-ph] (26 May 2020), y de Carlos Outeiral, Garrett M. Morris, …, Charlotte M. Deane, «Investigating the potential for a limited quantum speedup on protein lattice problems,» arXiv:2004.01118 [quant-ph] (02 Apr 2020).
Muchas gracias por tu comentario y por aclarar lo de la aplicabilidad de los ordenadores cuánticos para el plegado de proteínas. Y gracias también por las referencias, a las que echaré un vistazo seguro.