

Primero se logró la estructura 3D de la glicoproteína espicular S del coronavirus SARS-CoV-2 y su dominio de unión al receptor RBD (LCMF, 24 Feb 2020). Luego la del receptor de la célula huésped, la enzima convertidora de angiotensina humana hACE2 (LCMF, 07 Mar 2020). El siguiente paso era determinar la del complejo SARS-CoV-2 RBD/hACE2. Se publican dos artículos en Nature que la obtienen por cristalografía de rayos X, alcanzando sendas resoluciones de 2.45 Å y 2.68 Å. Cambios estructurales muy sutiles explican la mayor infectividad y patogénesis de SARS-CoV-2 (COVID-19) comparado con SARS-CoV (SARS); un par de puentes salinos y un puente de hidrógeno conducen a una unión más fuerte entre la cresta de los motivos de unión al receptor (RBM) de la proteína espicular del coronavirus y una hélice N-terminal de hACE2 en la membrana de la célula huésped. Sin lugar a dudas, lo que estamos aprendiendo sobre este coronavirus es muy relevante para el desarrollo de fármacos que combatan la COVID-19 y vacunas que controlen futuros rebrotes de la epidemia.
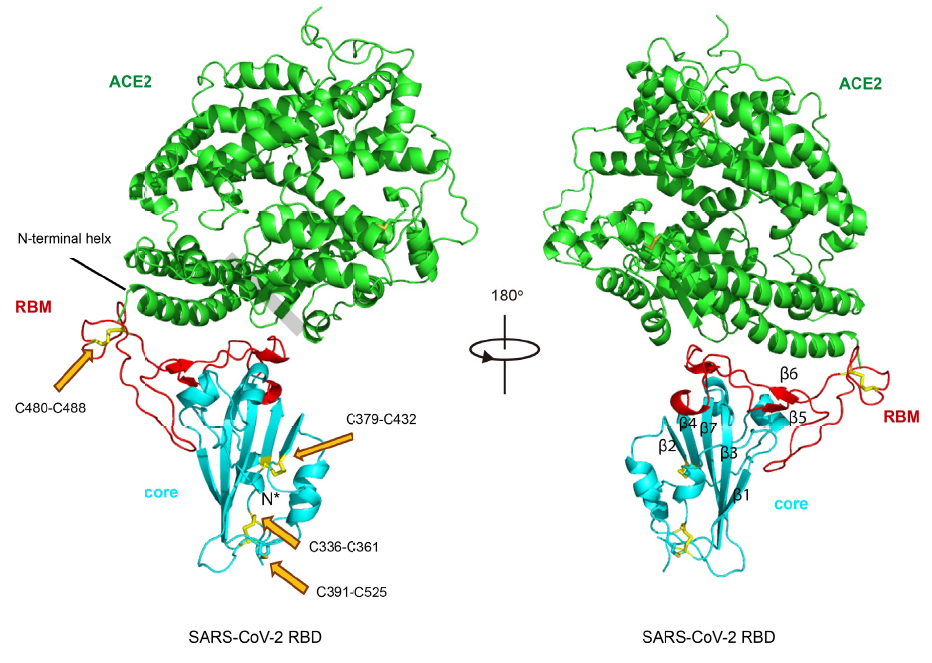
Los incautos pueden pensar que conocidas las estructuras de RBD y hACE2 inferir cómo se unen entre sí debe ser trivial (como juntar piezas de LEGO). Pero por desgracia la bioquímica estructural no es (un juego de niños) tan fácil. Se han realizado reconstrucciones in silico (mediante modelos teóricos usando ordenadores), pero la observación de la estructura cristalográfica real mediante difracción de rayos X es imprescindible. Por supuesto, el primer problema es como formar el complejo SARS-CoV-2 RBD/hACE2 con suficiente estabilidad para su observación; sin entrar en detalles, la experiencia previa en la formación del complejo SARS-CoV RBD/hACE2 (que se logró en 2005) ha sido clave (se aprovecha un puente salino entre Arg426 de RBD y Glu329 de hACE2 para reforzar la unión del complejo). En la figura se muestra en color rojo el RBM, en celeste el RBD y en verde hACE2; en amarillo, apuntados con flechas en naranja, se destacan los enlaces disulfuro; Cys336-Cys361, Cys379-Cys432 y Cys391-Cys525 estabilizan las cinco hojas beta (β1, β2, β3, β4 y β7), y Cys480-Cys488 es clave en la unión entre el la cresta del SARS-CoV-2 RBM y la hélice N-terminal de hACE2.
Dos artículos espectaculares, no solo por la belleza de la ciencia que muestran, sino por las implicaciones tan relevantes que prometen en la gestión de la pandemia. Lo sé, quizás te aburran estos temas, pero me apasiona la ciencia en acción, y la bioquímica estructural. Los artículos son Jian Shang, Gang Ye, …, Fang Li, «Structural basis of receptor recognition by SARS-CoV-2,» Nature (30 Mar 2020), doi: https://doi.org/10.1038/s41586-020-2179-y; Jun Lan, Jiwan Ge, …, Xinquan Wang, «Structure of the SARS-CoV-2 spike receptorbinding domain bound to the ACE2 receptor,» Nature (30 Mar 2020), doi: https://doi.org/10.1038/s41586-020-2180-5.
[PS 02 abr 2020] Recomiendo leer la pieza de Ignacio López-Goñi, «No, el coronavirus no se ha escapado de un laboratorio», microBIO, 02 abr 2020. «La naturaleza tiene suficientes recursos como para generar este y otros muchísimos virus. Promover otras hipótesis conspiranoicas sin base científica alguna es una irresponsabilidad. Es como afirmar que la culpa del coronavirus la tienen … Astérix y Obélix». [/PS]
[PS 04 ago 2020] Recomiendo leer a Xiaoli Xiong, Kun Qu, …, John A. G. Briggs, «A thermostable, closed SARS-CoV-2 spike protein trimer,» Nature Structural & Molecular Biology (31 Jul 2020), doi: https://doi.org/10.1038/s41594-020-0478-5, bioRxiv preprint 152835 (17 Jun 2020), doi: https://doi.org/10.1101/2020.06.15.152835. [/PS]
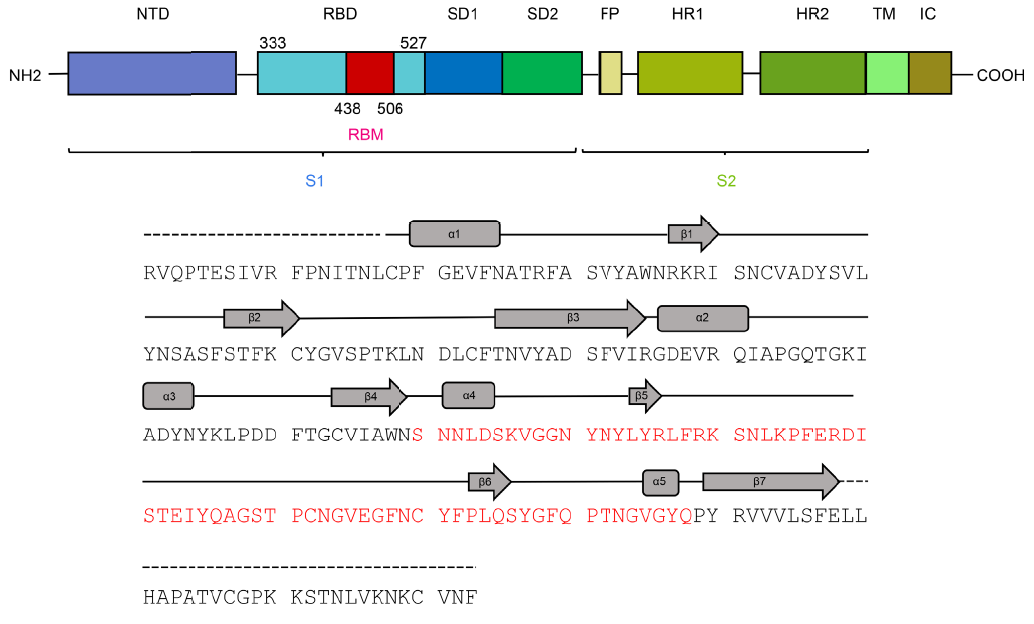
Esta figura muestra la estructrura primaria (secuencia de aminoácidos) del monómero de la proteína espicular S de SARS-CoV-2. El dominio terminal N (NTD) antecede al dominio de unión con el receptor (RBD) que incluye el motivo de unión con el receptor (RBM). Tras los subdominios SD1 y SD2, y el péptido de fusión (FP), aparecen las dos regiones de héptadas repetidas, HR1 y HR2. Finaliza la región transmembrana (TM) y el dominio intracelular (IC). Más detalles en mi pieza «La estructura 3D de la glicoproteína espicular del coronavirus SARS-CoV-2», LCMF, 24 feb 2020.

La comparación entre los complejos SARS-CoV RBD/hACE2 y SARS-CoV-2 RBD/hACE2 permite entender por qué es más infectiva COVID-19 que SARS. La SARS-CoV-2 RBM forma un interfaz de unión más grande y con más contacto con hACE2 que SARS-CoV RBM; una de las razones es que el puente salino entre SARS-CoV RBD y hACE2 es más débil que el de SARS-CoV-2 RBD y hACE2, pero más favorable energéticamente. Además, la estructura cristalina del complejo también contiene glicanos acoplados a los cuatro sitios de hACE2 y el sitio de RBD. El glicano acoplado a Asn90 de hACE2 forma un enlace he hidrógeno con Arg408 del núcleo de RBD; esta interacción se conserva entre SARS-CoV-2 y SARS-CoV.
Las diferencias estructurales entre las RBMs de SARS-CoV-2 y SARSCoV RBMs son sutiles, pero afectan a las conformaciones de los lazos en las crestas de unión al receptor (receptor-binding ridge en la figura). En ambas RBMs, uno de los lazos de la cresta contiene un enlace disulfuro que es crítico en la unión. SARS-CoV y bat-CoV Rs3367 contienen un motivo con tres residuos Pro-Pro-Ala en dicho lazo; pero en SARS-CoV-2 y bat-CoV RaTG13 muestran un motivo de cuatro residuos Gly-Val/Gln-Glu/Thr-Gly; así la conformación del bucle cambia gracias a que las glicinas son más flexibles. Este cambio favorece la unión RBD/hACE2. Además, la creta tiene una conformación más compacta gracias a los puentes de hidrógeno Asn487 y Ala475 en SARS-CoV-2 RBM, con lo que el lazo que contiene Ala475 se coloca más cerca de hACE2.
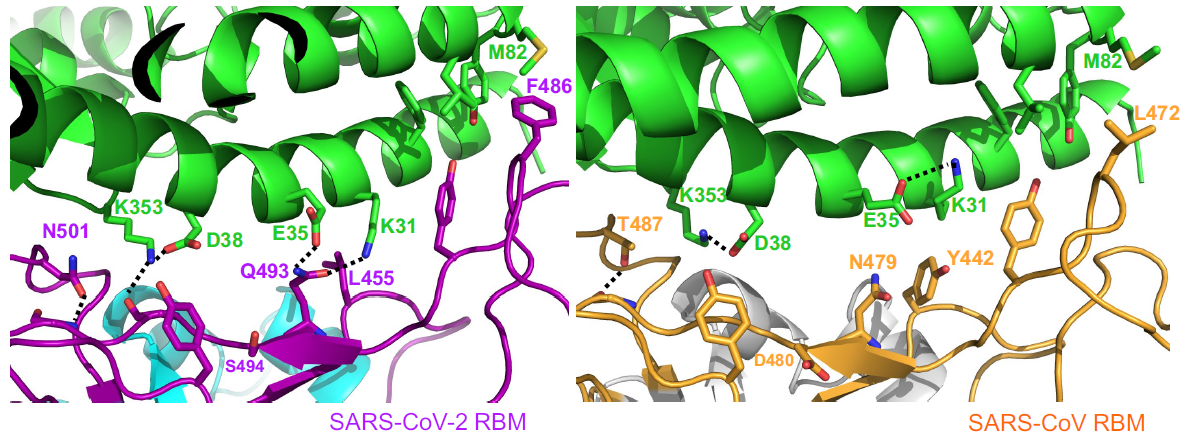
El contacto de la cresta de SARS-CoV-2 RBM con la hélice N-terminal de hACE2 es mayor que para SARS-CoV RBM. Por ejemplo, el residuo N-terminal Ser19 de hACE2 forma un nuevo enlace de hidrógeno con la cadena principal de Ala475 de SARS-CoV-2 RBM, y la Gln24 de la hélice N-terminal de hACE2 también forma un nuevo contacto con SARS-CoV-2 RBM. Al compararla con la Leu472 del SARS-CoV RBM, la Phe486 de SARS-CoV-2 RBM apunta en una dirección diferente y forma una región hidrófuga que involucra a Met82, Leu79, y Tyr83 de hACE2.
La comparación con SARS-CoV RBM muestra que estos pequeños cambios estructurales de SARS-CoV-2 RBM son más favorables para la unión con hACE2. Son diferencias sutiles, pero muy relevantes desde el punto de vista funcional. Se han desvelado dos puntos críticos de unión (virus-binding hotspots), el punto crítico hotspot-31 en el puente salino Lys31 y Glu35, y el hotspot-353 en otro puente salino entre Lys353 y Asp38. Estos dos puentes salinos son débiles, debido a la gran distancia en la interacción, pero como están encerrados en un entorno hidrófugo, que reduce la constante dieléctrica efectiva, su energía de enlace es mayor. Los artículos en Nature discuten este punto con bastante detalle (recomiendo a los lectores interesados una lectura detallada de los mismos).
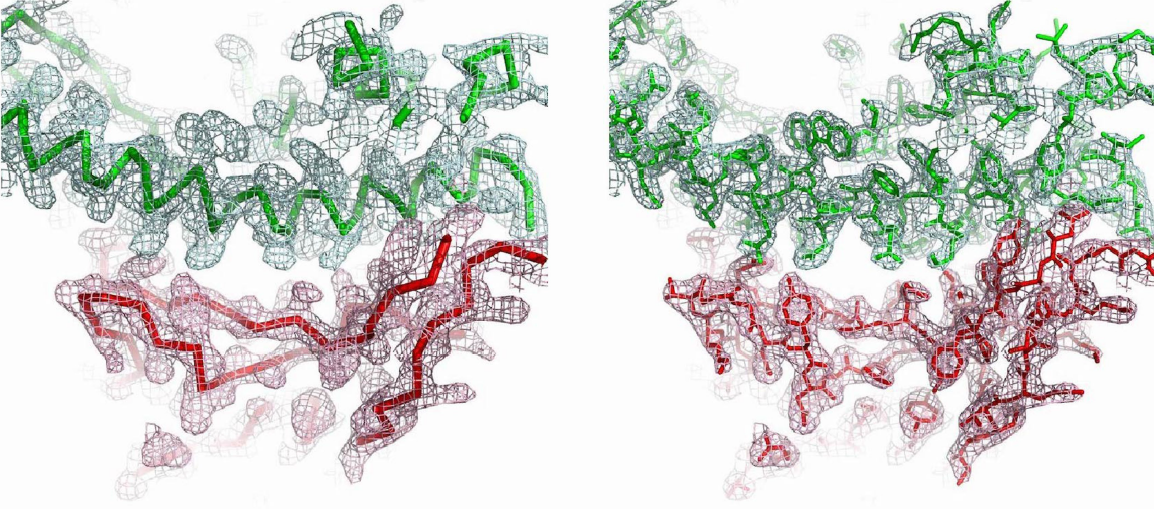
Esta figura muestra la densidad electrónica de la unión entre SARS-CoV-2 RBD (rojo) y hACE2 (verde). Las estructura del complejo permite desvelar por qué SARS-CoV-2 reconoce a hACE2 mucho mejor que SARSCoV. La cresta de unión de SARS-CoV-2 RBM es más compacta y forma mejor contacto con la hélice N-terminal de hACE2 gracias a cambios estructurales debidos al motivo de cuatro residuos (residuos 482-485: Gly-Val-Glu-Gly). Además, la Phe486 de SARS-CoV-2 RBM se inserta en una región hidrófuga, mientras la leucina correspondiente en SARS-CoV RBM forma un contacto más débil con hACE2 al tener una cadena lateral más pequeña. Más aún, los dos puntos críticos de unión (hotspots) son más estables al contener dos residuos de lisina acomodados en un ambiente hidrófugo que neutraliza sus cargas; los residuos Gln493 y Leu455 estabilizan el hotspot-31, mientras que Asn501 estabiliza el hotspot-353.

Para confirmar estas conclusiones estructurales se han realizado estudios bioquímicos para la afinidad de la unión RBD/hACE2 tras introducir ciertas mutaciones en SARS-CoV-2 RBD. No entraré en los detalles, pero destacaré que sugieren que el coronavirus de murciélagos RaTG13 podría infectar a los humanos (lo que ratifica el origen zoonótico de la epidemia). Además, las RBMs de SARS-CoV-2 y bat-COV RaTG13 contienen un motivo similar de cuatro residuos en la cresta de unión a ACE2, lo que apoya que uno ha evolucionado a partir del otro. Más aún, para mejorar el reconocimiento de hACE2, SARS-CoV-2 presenta dos cambios en los residuos L486F e Y493Q de RaTG13, que parece que han facilitado la transmisión de SARS-CoV-2 desde el murciélago a los humanos. Así, podría no haber un huésped intermedio entre el murciélago y el humano en la COVID-19, a diferencia de lo que ocurrió con SARS y con MERS. Por supuesto, por ahora es imposible descartar la existencia de un mediador, que bien podría ser un pangolín u otro animal salvaje vendido en el mercado de Wuhan; en el caso del pangolín es necesario secuenciar más genomas de coronavirus de pangolín para dilucidar la cuestión (algo que no están entre las prioridades de los científicos chinos ahora mismo).
En resumen, la ciencia sobre la infección COVID-19 y el coronavirus SARS-CoV-2 están avanzando a un ritmo asombroso. Los resultados que se están obteniendo son fascinantes. Y tendrán implicaciones clínicas en menos de un año. En un par de meses hemos logrado lo que costó varios años hace dos décadas. Y todo gracias a la ciencia básica. Porque lo que aprendes de una infección, como SARS o como MERS, que ya ni siquiera recordamos, acaba siendo de importancia capital en una nueva infección, como COVID-19. Esta pandemia nos recuerda que gastar en ciencia básica no solo es necesario, es imprescindible. Sin ciencia no hay futuro.
Hola, he entrado y te he conocido gracias a Julio Palacios, acabo de ver que sigues activo, ayer mismo, encontré este artículo https://jvi.asm.org/content/89/17/9119/article-info del 2015 del científico del que habla el vídeo de la RAI, qué opinas?
https://www.npr.org/sections/health-shots/2014/11/07/361219361/how-a-tilt-toward-safety-stopped-a-scientists-virus-research?t=1585662038989
Mil gracias
Sol, Ralph S. Baric es uno de los virólogos del comité de expertos que le dio nombre al nuevo coronavirus (que pasó de 2019-nCoV a SARS-CoV-2) https://www.nature.com/articles/s41564-020-0695-z y ahora está aplicando su trabajo sobre antivirales y vacunas para MERS a la nueva infección COVID-19.
En cuanto a la moratoria de 2014 sobre la investigación en los virus de la gripe, SARS y MERS, que cortó parte de la financiación pública en EEUU (pero no la de otros países, ni tampoco la privada) en este tema, acabó con un informe «Recommended policy guidance for potential pandemic pathogen care» (2016) sobre las recomendaciones de seguridad necesarias; por lo demás, por fortuna para los virólogos que ahora está liderando la investigación en COVID-19 no supuso un gran parón de financiación (como quizás pienses tras leer la noticia de NPR que citas).
En cuanto al artículo que citas, destaca la importancia del sitio de escisión S1/S2 en la proteína espicular S que diferencia entre el coronavirus de murciélago HKU4 y el de humanos MERS-CoV; expliqué su importancia en la fusión de las membranas del coronavirus y la célula huésped en mi pieza https://francis.naukas.com/2020/02/24/la-estructura-3d-de-la-glicoproteina-espicular-del-coronavirus-sars-cov-2/ «En los gammacoronavirus y en algunos betacoronavirus se produce una escisión de las subunidades S1 y S2 durante la fusión entre las membranas, mientras que en los alfacoronavirus y en algunos betacoronavirus no se produce dicha escisión. SARS-CoV-2 es un betacoronavirus en el que se produce la escisión,» como también en MERS-CoV (lo que afirma el artículo que citas). En los coronavirus humanos que producen resfriados no se produce dicha escisión.
Saludos
Francis
Gran artículo, Mula Francis, te has ganado mis respetos.
Insisto: me puedes decir si tienes un modelo de diseño de estructura o sólo haces «copy/paste». Gracias.
Ya contesté. Solo divulgo.
Muy bueno Francisco, excelente articulo…
Diferencias entre
cresta de los motivos de unión al receptor (RBM)
y y su dominio de unión al receptor RBD
En tu artículo ( puedo tutearte?) dies: «La proteína S es un trímero formado por tres péptidos, cada uno con dos subunidades S1 y S2.
La subunidad S1 actúa como una bisagra con dos conformaciones llamadas «abajo» (RBD down) y «arriba» (RBD up).
Pero ésto no es exactamente así en el vídeo que pones: o no lo he entendido.
Dentro video: https://youtu.be/cCwjLAR-0pk
No «visualizo» mas que variaciones sin que haya dos conformaciones abajo. Salvo error por mi parte.
La imagen por criomicroscopia electrónica muestra que solo uno de los péptidos está en estado «arriba», estando los otros dos en estado «abajo». La unión al receptor celular se realiza en la configuración «arriba». Tras la unión se escinden los tres péptidos de la proteína S por el punto S1/S2; luego se produce una segunda escisión por el punto S2′, que despliega el péptido de fusión (FP) clave en la unión entre las membranas.
Heinrich, la diferencia entre RBM y RBD es que RBM es un motivo, un trozo de secuencia, dentro de RBD (como muestra la figura https://francis.naukas.com/files/2020/03/D20200330-nature-s41586-020-2180-5-Overall-topology-SARS-CoV-2-spike-monomer.png
Puedes tuitearme. La acción de S1 como bisagra no está relacionada con las conformaciones abajo y arriba (lo que muestra el vídeo), sino a su acción tras la escisión de S1 y S2 en la fusión de membranas como ilustra esta figura https://francis.naukas.com/files/2020/02/D20200224-sinobiological-com-schematic-of-CoV-spike-protein-mediated-membrane-fusion.png de mi pieza previa https://francis.naukas.com/2020/02/24/la-estructura-3d-de-la-glicoproteina-espicular-del-coronavirus-sars-cov-2/
Saludos
Francis
Le supongo enterado de que para divulgar un artículo hay que pedir permiso a los autores. Se lo digo por las implicaciones legales. Le suena la EUIPO? A lo mejor soy abogado ( European Intelectual Property Office) ( mírelo en el buscador que use,) Ya se que Vd no lo hace con ánimo de lucro, y que no fusila o copia el artículo de Science, pero ojo que tiene que pedir los correspondientes permisos.
Se lo digo porque yo si he publicado en revistas de alto factor de impacto. Saludos.
https://s100.copyright.com/AppDispatchServlet#formTop
Gracias por informar. Yo también he publicado en revistas de alto factor de impacto.
Muy divertidas las aportaciones de Heinrich, sin entenderlas del todo me quedé con ganas de más.
Al respecto de lo que dice de aportar en una revista de alto impacto, me ha hecho preguntarme qué es más importante, si un gran cientifico o un gran divulgador. ¿Ha hecho, por poner ejemplos, más por la ciencia Kip Thorne que Carl Sagan?
Uno ha hecho una aportación directa y el otro una inversión para tener futuros cientificos y una sociedad con conocimientos en ciencia. Creo, Heinrich, que no se debería de infravalorar la divulgación.
De la otra faceta no conozco, pero gracias por tu labor divulgativa Francis, te sigo desde hace tiempo en tu página y más todavía cuando apareces en Coffee Break, que entiendo algo más.
Un saludo
Por supuesto que
1, El artículo de divulgacion el Dr Francisco Villatoro es excelente.
2. Deja muy claro que hace divulgación, muy amena, y no simplemente fusilar artículos .
3. Yo tengo hasta ahora,sobre 30 publicaciones. nunca me importa que me citen, si dejan claro de donde viene el material. Y Mula Francis ( con respeto) lo dice y deja los doi. Nada que decir.Nada que objetar.
La verdad es que en AAAS y en New York Academy of Sciences yo estuve suscrito muchos años como socio o member, en realidad solo querían cobrar la cuota y las menciones a ls artículos e hacen mas por cortesía o por que la revista a donde envíes tu trabajo te lo exija y los referees hagan su trabajo.
Una duda: Dice en el texto
«Por supuesto, el primer problema es como formar el complejo SARS-CoV-2 RBD/hACE2 con suficiente estabilidad para su observación; sin entrar en detalles, la experiencia previa en la formación del complejo SARS-CoV RBD/hACE2 (que se logró en 2005) ha sido clave (se aprovecha un puente salino entre Arg426 de RBD y Glu329 de hACE2 para reforzar la unión del complejo)»
Perecto..
Luego especifica como los diversos colores pertenecen el rojo al RBM ,etc…y los puentes disulfuro que los señala con una flecha naranja. pero das solo trs pareja sy luego hablas de ; Cys336-Cys361, Cys379-Cys432 y Cys391-Cys525 estabilizan las cinco hojas beta (β1, β2, β3, β4 y β7),
Bien…pero luego me metes esta frase que se refiere al SARS-Cov-2
» se destacan los enlaces dy Cys480-Cys488 es clave en la unión entre el la cresta del SARS-CoV-2 RBM y la hélice N-terminal de hACE2.»
Me he perdido, estás hablando d ela estructira de union establecida en 2005 para el Co-1, peor luego me metes los del SARS-CoV-2. ? La estructura que pones es del SARS-CoV2, creo, luego no se si es una traducción literal ote estas refiriendo TODO EL TIEMPO al SARS-CoV-1.
Saludos.
Solo hablo de SARS-CoV-2, pero para cristalizar el complejo fue clave la experiencia previa con SARS-CoV. Quizás no está bien escrito. Puedes leer el artículo original para leerlo de primera mano, es gratis (a veces traduzco varios párrafos en un par de frases y puede que se entienda mejor leyendo los párrafos originales).
Es increíble lo que se puede conseguir hoy en día con los grandes súper ordenadores militares y sus cientos de petaflops, calcular todas estas sutilezas estructurales a base de cambiar una letra o dos en el código aquí y allá y probar todas las combinaciones y simular las interacciones es un trabajo de computación gigantesco.
Bravo, se acerca el momento en el que el trabajo de cálculo necesario para crear un antiviral o una vacuna esté al alcance de cualquiera, con lo cual se acabarán todos estos laboratorios militares de nivel 4 y todo el mundo dispondrá de más tiempo de computación libre en los ordenadores más rápidos del mundo.
Es un poco cándido pensar que teniendo un virus con el del Sars y con papers publicados desde hace años alertando de lo fácil que sería que con una modificación ALEATORIA se volviese más virulento no iba a haber gobiernos petando los petaflops en calcularlo, si estaba a huevo, señores.
No es casualidad que intercalado en esta misma serie de estructura de proteínas y receptores Francis nos haya obsequiado con otro artículo dedicado a las IA barriendo bases de datos de moléculas para calcular la que es más afín al virus y puede servir de antiviral, es el mismo juego pero versión «pobre» (y versión buena, obviamente).
Francis dos preguntas
1. seria necesario usar SLAC para ver la dinamica de esta union para lograr vacunas o antivirales? El antiviral que termina su fase 3 en abril es prometedor el de Gilead?
2. el resto de la proteina no incide en la union? las fuerzas electromagneticas son solo de tan corta distancias las que influyen en la union y dinamica?
Benjamin, (1.1) el láser de rayos X de SLAC se usa para hacer cristalografía de rayos X; no es posible ver la «dinámica» solo obtener «imágenes estáticas» a partir de las cuales (en ciertos casos) se puede inferir la dinámica. En los artículos de Nature se han usado el APS (Advanced Photon Source) del ANL (Argonne National Laboratory), EEUU, y la instalación del Technology Center of Protein Research en la Tsinghua University, Beijing, China. (1.2) El antiviral remdesivir para el tratamiento de COVID-19, que está ensayando en fase 3 la empresa biotecnológica Gilead Sciences, es prometedor in vitro, pero hay que esperar a las conclusión de los ensayos in vivo. También se está usando en muchos otros ensayos, hay que esperar a sus conclusioes.
(2.1) Las proteínas tienen sitios activos donde se realiza la función biológica. Gracias a ello pueden mutar otras partes de la proteína (en los virus de ARN ocurre mucho) sin que se afecte a su función biológica. (2.2) En las uniones por puentes salinos y puentes de hidrógeno las fuerzas intermoleculares relevantes son electrostáticas (interacción entre dipolos), siendo fuerzas débiles y de corto alcance (si no has leído nunca nada sobre ellas, empieza por la wikipedia https://es.wikipedia.org/wiki/Fuerza_por_puente_de_hidr%C3%B3geno ).
Saludos
Francis
Hola francis
Que pasa despues de levantarse la cuarentena? se vuelve al punto anterior al ponerla? entonces la curva se vuelve a repetir hasta la vacuna?
Muchas gracias por las aportaciones.
Creo que este hallazgo (si se confirma experimentalmente) ayudaría a explicar la alta contagiosidad de este virus, su capacidad replicativa en faringe mil veces superior al SARS, con el concepto añadido de «portadores asintomáticos» contagiadores, así como la aparente baja tasa de infecciones en la infancia (pero con potencial de infectar a adultos). Habría entonces qué revisar el concepto del ACE2 cómo único receptor del virus por las células del huésped. Quizá haya una sinergia entre integrinas (fijar y mantener el virus en vías altas) y ACE2 (propiciar la internalización, sobre todo en pulmón). En este sentido, pudiera ser qué la expresión de ACE2 sea muy baja en los niños y vaya aumentando con la edad. Es otra hipótesis a considerar.
Un cordial saludo.
«Dos artículos espectaculares, no solo por la belleza de la ciencia que muestran, sino por las implicaciones tan relevantes que prometen en la gestión de la pandemia. Lo sé, quizás te aburran estos temas, pero me apasiona la ciencia en acción, y la bioquímica estructural.»
Mi respuesta, no no me aburren en absoluto, un amagnífico artículo,deliciosamente bien contado.
Mula Francis dijo:
«La comparación entre los complejos SARS-CoV RBD/hACE2 y SARS-CoV-2 RBD/hACE2 permite entender por qué es más infectiva COVID-19 que SARS.»
Heinrich Böll pregunta:
¿que relacción existe entre tener una R0 de 2,5 y la » La SARS-CoV-2 RBM forma un interfaz de unión más grande y con más contacto con hACE2 que SARS-CoV RBM;» que relacciona la interfaz? ¿ que el COVID-19? tiene mas » superficie» para acoplarse al receptor ACE/2 ?
Mula Francis dice:
«una de las razones es que el puente salino entre SARS-CoV RBD y hACE2 es más débil que el de SARS-CoV-2 RBD y hACE2, pero más favorable energéticamente. Además, la estructura cristalina del complejo también contiene glicanos acoplados a los cuatro sitios de hACE2 y el sitio de RBD. El glicano acoplado a Asn90 de hACE2 forma un enlace he hidrógeno con Arg408 del núcleo de RBD»
Bueno, estoy es Korolkovas hecho magia. pero insisto: esto no puede extraplarse a la MORTALIDAd, ese es un concepto clínic que dependerá como está el huesped. Con lo de la infectividad quieres decir que según esté mas o emnos estable energéticamente se va a unir el COVID-19 con más facilidad? ¿que hay de cierto en eso de ue hace falta un «residuo de furilo» ( eso no es de tu artículo, lo he leído en la Academia de medicina Mexicana: link:https://www.youtube.com/watch?v=I0AbpnFP1g8 dicen que «un furilo» es necesario para «anclar» al virus en el receptor hACE/2. Eso me ha dejado descolocado, proque no dan la exhaustiva información que tú das en tu artículo….
Con todo lo que sabes de MATEMATICAS Y FISICA ¿CÓMO ES QUE NO TE ESTÁN FICHANDO PARA UN EQUIPO DE INVESTIGACIÓN? Madre mia, que desperdicio…
Heinrich, el R0 depende de la infección, no del virus. No te confundas. Además, su valor depende del modelo matemático de la epidemia que se use; por ello se calcula a posteriori, tras el final de la epidemia, usando el modelo que mejor describa su evolución. Su relación con la bioquímica de la unión del virus a la célula diana es muy, pero muy indirecta.
Hola y buenas tardes. Muy interesante su artículo, muy buen ejemplo de lo que es divulgación científica acertada.
Tengo una par de dudas, si Vd logra satisfacer mi curiosidad, le quedaré muy agradecido:
Vd. dice que : «La comparación entre los complejos SARS-CoV RBD/hACE2 y SARS-CoV-2 RBD/hACE2 permite entender por qué es más infectiva COVID-19 que SARS. La SARS-CoV-2 RBM forma un interfaz de unión más grande y con más contacto con hACE2 que SARS-CoV RBM;» es decir, relacciona una estructura puramente bioquímica con un interfaz de unión más grande, con un concepto clínico, como es la infectividad. Intenta decir que esa unión del SARS-CoV-2 al receptor angiotensina es mucho mas fuerte y eso hace que sea más infectivo o ¿que se transmita mejor, porque la unión es más estable, mas fuerte?.
Y no podría ser que de un genoma del murcielago , con un 96 % de similitud de forma exógena se » implante» una proteína S? Con las tecnicas de Sanchez Mogica de tecnologia CRIPSR cas9 no es descabellado… Lo dice una teoria, una de las muchas teorias conspirativas. Yo no creo en ellas, pero…cuantos más datos aparezcan, mejor lo sabremos.
Francis, lo dice tú en tu artículo, «La comparación entre los complejos SARS-CoV RBD/hACE2 y SARS-CoV-2 RBD/hACE2 permite entender por qué es más INFECTIVA COVID-19 que SARS.»
Me podria decir y explicar como influye la proteína de la hélice N-terminal de la hACE2(angiotensina) de la célula huésped, la proteína espicular S del SARS-CoV-2 y la capucha metilada en el extremo 5′ y una cola poliadenilada poli-A en el extremo 3‘ del ARN monocatenario del COVID-19 para que sea 10 veces más letal e infeccioso por mutación controlada que la gripe común.
Hola¡¡
Sólo agradecerte por tu valioso y gran trabajo.. Es real ayuda, para poner tener un revisión científica y entretenida del tema¡¡..
Saludos desde Chile
Buenas, he estudiado varios papers del 2020 que tratan la unión del la spike del virus con la ACE2 humana, especialmente el paper de Nature de LAN 2020 (Lan, J., Ge, J., Yu, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature (2020). https://doi.org/10.1038/s41586-020-2180-5).
Estaba buscando algún resultado en el que una variación de aminoácidos entre la spike del coronavirus SARS-CoV-2 con respecto al SARS-CoV le diese una potencial ventaja en el (hipotético) uso por parte del virus de la transglutaminasa humana para facilitar la unión o entrada a la ACE2 de la célula huèsped.
Me ha sorprendido ver una variación de uno con respecto a otro en la K417 (SARS-CoV-2) frente a la R426 (SARS-CoV) en la spike de ambos (resíduo que está en el RBD pero no en el RBM) para unirse a la Q24 de la ACE2 humana.
Esto se puede ver sólo en la tabla Extended Data Table 2 del citado estudio, en todos los otros estudios que he buscado del 2020 en TODOS dice que la K417 del SARS-CoV-2 de une a una D30 en la ACE2 y no a una Q24 como dice esa tabla.
No soy muy ducho en esto, pero me podría aclarar si hay un fallo en este trabajo de Nature o realmente la variación hacia K417 de la spike del SARS-CoV-2 es la que se une a la Q24 de la ACE2 como parece citar el trabajo de LAN 2020 en la citada tabla «Extended Data Table 2»?
Saludos
FOTGCREN
https://www.youtube.com/channel/UCaicdliA5suAqr9nA3rIapg
Exactamente lo que pido que me aclare alguien que sepa, es por que poniendo esto en el texto del artículo:
LAN 2020 «Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor»
https://www.nature.com/articles/s41586-020-2180-5
«Outside the RBM, there is a unique ACE2-interacting residue (Lys417) in SARS-CoV-2, which forms salt-bridge interactions with Asp30 of ACE2 (Fig. 3b). This position is replaced by a valine in the SARS-CoV RBD that fails to participate in ACE2 binding (Figs. 2b, 3b). Furthermore, a comparison of the surface electrostatic potential also identified a positive charged patch on the SARS-CoV-2 RBD contributed by Lys417 that is absent on the SARS-CoV RBD (Fig. 3b). These subtly different ACE2 interactions may contribute to the difference in binding affinity of the SARS-CoV-2 and SARS-CoV to the ACE2 receptor (4.7 nM compared with 31 nM, respectively) (Extended Data Fig. 4).»
O sea, que hay una lisina (K417) en el spike del SARS-CoV-2 y no en el spike del SARS-CoV (que tiene una valina V en su lugar) que interacciona con una asparagina (D30) en el hACE2 que parece darle ventaja en la unión…. Pero luego en la «Extended Data Table 2» (Contact residues of the SARS-CoV-2 RBD–ACE2 and SARS-CoV RBD–ACE2 interfaces) pone lo primero que K417 es un contact residue con Q40 en hACE2, lo cual es lo que a mi me interesa, ya que para una hipotética participación de la transglutaminasa humana en el evento es necesaria la presencia de un resíduo glutamina Q y un resíduo lisina K que estén en proximidad…..
La tabla que cito (Extended Data Table 2) esta ahi:
https://www.nature.com/articles/s41586-020-2180-5#Sec10
https://www.nature.com/articles/s41586-020-2180-5/tables/3
pero no se ve y hay que verla en el pdf del articulo:
https://www.nature.com/articles/s41586-020-2180-5.pdf
¿alguien me puede explicar esto?
¿K417 en la spike de SARS-CoV-2 y Q24 en la hACE2 contactan en la unión de la spike del virus SARS-CoV-2 con el receptor hACE2 de la célula huesped en el proceso de adhesión?
gracias
FOTGCREN
https://www.youtube.com/channel/UCaicdliA5suAqr9nA3rIapg
Buenas tardes,
el hecho de que COVID-19 sea más patogenico que SARS se debe unicamente a 2 puentes salino más y un puente de hidrogeno? O es una forma de hablar?
Gracias
Como debería ser obvio, Ainhoa, influyen muchísimos factores, aunque solo conocemos algunos de ellos. Comparándolo con SARS-1, destaca que la unión entre la proteína espicular de SARS-2 con la proteína (receptor) ACE-2 (o ECA-2 en español) es más fuerte, con lo que el virus se puede unir a varias ACE-2 facilitando su endocitosis; es más fuerte gracias a esos puentes salinos y de hidrógeno. También que la proteína espicular presenta un sitio de escisión S1/S2 que permite la acción de la furina habilitando un segundo mecanismo de infección, la fusión de membranas. Pero influyen muchos otros factores.
Hola, Francisco.
Yo digo que el agravamiento de las personas con condiciones de base relacionadas a una mala evolución, tienen un común denominador, que puede ser los altos niveles de beta 2 microglobulina. Pienso que esta proteína humana tiene mucha similitud con un péptido viral de carácter antigénico. Sobre todo se parecen cuando la B2M cambia su conformación para el transporte de fosfolípidos como la cardiolipina. Es tal el parecido entre el péptido viral y el humano, que el sistema inmunológico responde de manera autoreactiva y ahí es que se desencadena todo lo que ya se sabe. Por eso las complicaciones aparecen después de 10 días a 2 semanas, coincidiendo con la elevación de anticuerpos. Los clínicos siempre empiezan el cuento a partir de la «tormenta de citoquinas» y el «síndrome antifosfolipídico», pero eso no sucede de la nada, hay que ir más atrás. Eso definiría los protocolos (no hablaré de esto ahora, comprendo que no es tu área). Yo he estado mirando la B2M y ese tramo del RBD en S, entre C379 y C432, se me parecen bastante y ambas están muy glicosiladas. ¿Tienes cómo comparar estos dos péptidos, por favor?
Saludos,
Patricia Jiménez (médica, viróloga)
Patricia, no entiendo qué quieres exactamente, ¿comparar las estructuras primarias de esta proteína https://www.ncbi.nlm.nih.gov/protein/184210 con la de esta proteína https://www.ncbi.nlm.nih.gov/protein/1796318598? ¿Comparar las estructuras secundarias? ¿Las terciarias? ¿Realizar un docking entre ambas?
Son proteínas que se parecen muy poco (B2M tiene 338 aa. y S tiene 1273 aa.).
Un alineamiento de la subsecuencia que comentas S(379:432) con toda B2M usando la matriz PAM250 ofrece B2M(171:227) = S(379:432)
B2M = CEAANVAEQRRAYLEGTC-VE-WLHRYLENGKEMLQRADPPKTHVTHHPVFDYE-AT-LR-C
-S- = C-YG-VS-PTK–LNDLCFTNVYADSFVIRGDEV–RQIAP-GQTGKIADYNYKLPDDFTGC
Un alineamiento de la subsecuencia que comentas S(379:432) con toda B2M usando la matriz BLOSUM45 ofrece B2M(171:227) = S(379:432)
B2M = CEAANVAEQRRAYLEGTCVEWLHRYLENGKEMLQRADPPKT-HVT-H-HPVFDYEATLRC
-S- = CYG–VSPTKLNDLCFTNV-YADSFVIRGDEVRQIA-PGQTGKIADYNYKLPD-DFT-GC
Un alineamiento local usando la matriz PAM250 ofrece B2M(12:232) = S(877:1212)
B2M = LLSGALTLTETWA-GS-HSMRY-FSAAVS-R-PGRGEPRFIAMGYVDDTQFVRFDSDSAC
-S- = LLAG–TITSGWTFGAGAALQIPFAMQMAYRFNGIGVTQNV-L-Y-ENQKLIANQFNSAI
B2M = PRMEPRAPWVEQE-GPEYWEEETRNTKAHAQTDRMNLQTLRGYYNQSEAS-SHTLQWM–
-S- = GKIQDSLSSTASALG-KL-QDVVNQ-NAQAL-NTL-VKQLSSNFGAISSVLNDILSRLDK
B2M = IGCDLGSDGRLLRG-YEQY-AYDGKDYLALNEDLRSWTA-ADTA-AQ-I—SKR-K-CE
-S- = VEAEVQID-RLITGRLQSLQTYVTQQ-LIRAAEIRASANLAATKMSECVLGQSKRVDFCG
B2M = AA-NVAE-QRRAYLEGTCVEWLH-RYL-ENGKEML-QRA–DPPKTHVTHHPVFDYEATL
-S- = KGYHLMSFPQSA-PHGV-V-FLHVTYVPAQEKNFTTAPAICHDGKAHFPREGVFVSNGT-
B2M = RCWALG—FYPAEIILTWQRD-GEDQTQDVELVETRPAGDGTFQKWAAVVVPSGEE-QR
-S- = H-WFVTQRNFYEPQIITT-DNTFVSGNC-DV-VIGI–VNNTVYDPLQPELDSFKEELDK
B2M = Y—-TC-HVQH-E–GLPEPLM-LRWKQ-SSLPTIPIMGIVAGLVVLAAVVTGAAVAAVLW
-S- = YFKNHTSPDVDLGDISGINASVVNIQ-KEIDRLNEVA-KNLNESLIDLQEL–GKYEQYIKW
Un alineamiento local usando la matriz BLOSUM45 ofrece B2M(3:323) = S(869:1231)
B2M = VMAPRTLFLLLSGALTLTETWA-GSHS-MRY-FSAAVS-R-PGRGEPRFIAMGYVDDTQF
-S- = MIAQYT-SALLAG–TITSGWTFGAGAALQIPFAMQMAYRFNGIGVTQNV-L-Y-ENQKL
B2M = VRFDSDSACPRMEPR-APWVEQEGPEYWEEETRNTKAHA-QTDRMNLQTLRGYYNQ–SE
-S- = IANQFNSAIGKIQDSLSSTASALG-KL-QD-VVNQNAQALNTLVKQLSSNFGAISSVLND
B2M = ASSHTLQWMIGCDLGSDGRLLRGYEQ-Y-AYDGKDYLALNEDLR-SWT-AADTAAQ-I–
-S- = ILSR-LD-KVEAEVQID-RLITGRLQSLQTYVTQQLIRAAE-IRASANLAATKMSECVLG
B2M = -SKR-K-C-EAANVAE-QRRAYLEGTCVEWLH-RYLENGKEM-LQRADPPKTH–VTHHP
-S- = QSKRVDFCGKGYHLMSFPQSAP-HGV-V-FLHVTYVP-AQEKNFTTA-PAICHDGKAHFP
B2M = —VFDYEATLRCWAL—GFY-PAEIILT—W-Q-R-D—G–E—-D–QTQ-D-
-S- = REGVFVSNGT-H-WFVTQRNFYEP-QIITTDNTFVSGNCDVVIGIVNNTVYDPLQPELDS
B2M = V-E-L—VE–TRPAGD-GTFQKWAAVVVPSGEE-QRYTCHV-QH–EGLPEPLM-L-R
-S- = FKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLN-EVAKNLNESLID-LQELGK
B2M = WKQS-SLPTIPI-MGIVAGLVVLAAVVT
-S- = YEQYIKWPWY-IWLGFIAGLIAIV-MVT
Si lo copias y lo pones en letra tipo Courier lo verás mejor (más alineado).
No sé si te servirá de algo.
Saludos
Francisco
Te felicito por tu extraordinaria explicación y la capacidad de hacerte entender sobre algo del mecanismo fisiopatológico a nivel molecular para que podamos entender porque este virus tiene esa capacidad de infectar tan eficientemente.
Espero que usted se encuentre bien de salud. Con el debido respeto sólo divulgue la información científica y no emita ningún otro comentario.
«La naturaleza tiene suficientes recursos como para generar este y otros muchísimos virus. Promover otras hipótesis conspiranoicas sin base científica alguna es una irresponsabilidad. Es como afirmar que la culpa del coronavirus la tienen … Astérix y Obélix»
Entonces me pregunto ¿Será la actual pandemia de COVID un NUEVO mecanismo de regulación de la especie o una nueva forma de eliminar a los opositores?
Cada vez hay mas evidencia científica, que sostiene que el coronavirus SARS-CoV-2, es hecho por los humanos y NO por la naturaleza.
https://www.infobae.com/america/mundo/2020/09/15/una-virologa-desertora-china-publico-un-estudio-que-afirma-que-el-coronavirus-fue-creado-en-un-laboratorio/?fbclid=IwAR37TVnXOHKN0wvO-QvSg7TzFFG20etwKagNHErDmVA0DQh7FajEDVlNkOc
Cabazorro, el «artículo» de Li-Meng Yan y sus colegas se ha «publicado» en la «prestigiosa» web Zenodo (porque fue «strictly censored on peer-reviewed scientific journals»): https://zenodo.org/record/4028830. Todos los autores están afiliados a la «prestigiosa» institución Rule of Law Society & Rule of Law Foundation, New York, NY, USA (https://www.rolsociety.org/, visítala para hacerte una idea si no la conocías ya). El «artículo» no tiene ni pies ni cabeza. No es un artículo científico. No es ciencia. ¿Qué es? Te recomiendo leerlo para hacerte tu propia opinión.