


Las técnicas de edición genética CRISPR-Cas9 han revolucionado la ingeniería genética en células eucariotas. Su gran problema son las mutaciones no deseadas (off-target, o «fuera del objetivo»). El futuro de estas técnicas es el diseño del ARN guía apropiado para cada paciente. Se han publicado en Science tres artículos que presentan métodos para identificar las mutaciones no deseadas, con objeto de evaluar la efectividad de la edición de genes en un paciente concreto, así como de los editores de bases. Estas técnicas in vitro examinan el genoma de las células que serán editadas, determinando los posibles lugares off-target y permitiendo el diseño personalizado del ARN guía para minimizar sus efectos colaterales. Sin lugar a dudas un gran avance para el uso terapéutico de la edición genética.
Un artículo presenta la nueva técnica llamada DISCOVER-Seq (discovery of in situ Cas off-targets and verification by sequencing), para el descubrimiento in situ de las mutaciones no deseadas debidas a endonucleasas Cas y su verificación mediante secuenciación. En los lugares donde actúa Cas9 se corta el ADN y la célula activa mecanismos de reparación. La idea es localizar estos lugares usando la maquinaria de reparación de los cortes del ADN presente en la propia célula (en concreto una proteína de reparación llamada MRE11). El método funcionó con éxito en células madre pluripotentes inducidas (iPSCs) de un paciente con síndrome de Charcot-Marie-Tooth, así como en hígados de ratón que se editaron con Cas9 administrado por adenovirus.
Los otros dos artículos muestran en ratones y en el arroz que los editores de bases de adenina producen menos mutaciones no deseadas que los de citosina. En el libro de Lluís Montoliu “Editando genes: recorta, pega y colorea» (LCMF, 13 Feb 2019), los editores de bases son la parte «colorea» del título. Estas técnicas CRISPR permiten cambiar una sola base, es decir, introducir o corregir una mutación de tipo SNP (polimorfismo de un solo nucleótido); por ejemplo, C(itocina) por T(imina), o A(denina) por G(uanina). Los nuevos artículos muestran que el editor de bases de citosina (BE3) genera unas 20 veces más variantes de un solo nucleótido no deseadas que el editor de bases de adenina (ABE7). La mayoría de las mutaciones no deseadas (off-target) son independientes de la guía de ARN, lo que implica que su posición no fue debida a un error de la dCas9, sino de la actividad aleatoria no deseada de la desaminasa fusionada a la dCas9 en el editor de bases.
Los tres artículos son Beeke Wienert, Stacia K. Wyman, …, Jacob E. Corn, “Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq,” Science 364: 286-289 (19 Apr 2019), doi: 10.1126/science.aav9023 [web], bioRxiv:10.1101/469635; Erwei Zuo, Yidi Sun, …, Hui Yang, “Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos,” Science 364: 289-292 (19 Apr 2019), doi: 10.1126/science.aav9973 [web]; Shuai Jin, Yuan Zong, …, Caixia Gao, “Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice,” Science 364: 292-295 (19 Apr 2019), doi: 10.1126/science.aaw7166 [web]. A nivel divulgativo recomiendo Hannah R. Kempton, Lei S. Qi, “When genome editing goes off-target,” Science 364: 234-236 (19 Apr 2019), doi: 10.1126/science.aax1827 [web].
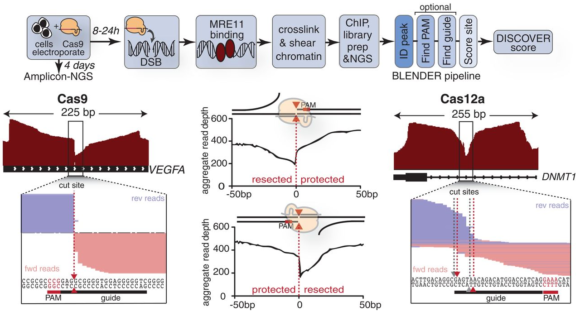
Desde su invención en 2012, para evitar las mutaciones no deseadas, se diseña el ARN guía mediante un software bioinformático que usa un genoma de referencia. Pero estas técnicas in silico tienen un problema, porque el genoma de un paciente difiere del genoma humano de referencia, con lo que el ARN guía puede acabar en un lugar inapropiado que no ha sido predicho in silico. El artículo liderado por Jacob E. Corn, del Innovative Genomics Institute (IGI), University of California Berkeley, cuyos primeros autores son Beeke Wienert y Stacia Wyman, tras estudiar varias proteínas de reparación del ADN, encontraron que la proteína reparadora de doble hebra (MRE11, recombinación meiótica 11) es una de las primeras en responder a un corte. Su idea es identificar las ubicaciones donde la proteína MRE11 está insertada en el ADN y secuenciar las regiones adyacentes para determinar las posibles mutaciones no deseadas (todos los off-targets).

Para la secuenciación de un genoma completo se usa una técnica de rotura del ADN en trozos pequeños (como la sonicación por ultrasonidos), una técnica de amplificación de ADN (que incrementa el número de copias de cada trozo) y una técnica de reconstrucción basada en los solapes entre trozos. Si el ADN tiene insertadas proteínas MRE11, habrá un gran número de trozos que acaban o empiezan en dichos lugares. Así, un diagrama del número de solapes mostrará un exceso de estos trozos, que permitirá identificar su posición con una gran precisión. En esta figura se consideran trozos de 100 pares de bases y se observan cambios bruscos en los lugares donde se encuentra el corte (cut site) y en los extremos de los trozos que lo determinan. La técnica DISCOVER-Seq usa una modificación del software CHIP-Seq para determinar las mutaciones no deseadas (el nuevo software se llama BLENDER).
La nueva técnica tiene un gran abanico de aplicaciones en la edición genética con fines terapéuticos. La más importante es que permite el diseño personalizado guías de ARN que minimicen las mutaciones no deseadas para un paciente concreto (sobre todo en los casos de enfermedades raras asociadas a un único SNP). El análisis ex vivo antes del tratamiento genético proporciona un nivel adicional de seguridad para las terapias génicas que usen endonucleasa CRISPR-Cas. Sin lugar a dudas, un gran avance en el campo de las técnicas de edición genética.
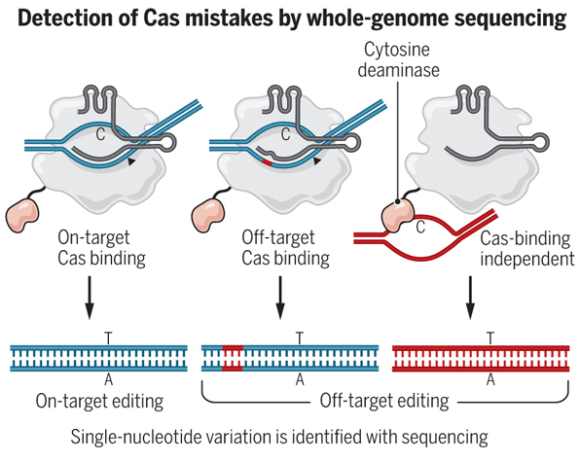
Las proteínas Cas (asociadas a secuencias CRISPR) son endonucleasas que cortan las dos cadenas del ADN en un lugar específico del genoma dictado por una pequeña molécula de ARN (llamada ARN guía). Los editores de bases usan las tijeras Cas9 (catalíticamente) inactivadas, la llamada Cas9 muerta (dCas9, dead Cas9), con una doble mutación que las hace incapaces de cortar el ADN, pero sirven para localizar y marcar una posición del genoma con gran precisión mediante el ARN guía. La proteína dCas9 se queda enganchada en dicha posición; si logramos que la proteína Cas9 transporte otra proteína, dicha proteína actuará en dicho lugar, por ejemplo, una proteína fluorescente. Pero también se puede producir el cambio de una base por otra, para ello se fusiona con la enzima Cas una proteína de tipo desaminasa de nucleótidos. Esta herramienta no genera roturas de doble cadena en el ADN, sino que utiliza la desaminasa para convertir un nucleótido en otro en la posición seleccionada. En ambos casos, es fundamental contar con un método apropiado para identificar la actividad fuera del objetivo para la herramienta dada.
En el editor de bases BE3 se une a la dCas9 una desaminasa de la citosina que al encontrar la pareja de bases CG, cambia la C por una U, dando UG; la maquinaria celular repara este error sustituyendo la U por una T, dando lugar a TG y luego repara el error de apareamiento TG sustituyendo la G por una A, dando lugar a TA; así que de CG se pasa a TA. En el editor de bases ABE7 se acopla a dCas9 una adenosina desaminasa que promueve la desaminación de la adenosina, dando como resultado inosina; así se transforma AT en IT, que se repara dando GT que a su vez se corrige a GC, luego se logra cambiar AT por GC.

El segundo artículo en Science está liderado por Hui Yang, del Institute of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, siendo un trabajo conjunto de sus primeros cuatro autores Erwei Zuo, Yidi Sun, Wu Wei y Tanglong Yuan, habiendo colaborado investigadores del Stanford Genome Technology Center, Stanford University. Se ha desarrollado una técnica llamada GOTI (genome-wide off-target analysis by two-cell embryo injection, análisis de mutaciones no deseadas en todo el genoma mediante la inyección en embriones de dos células) para identificar in vivo mutaciones no deseadas en los editores de bases de citosina y adenina en embriones de ratón. Han comparado los editores de bases de la citosina BE3 (base editor 3) y de la adenina ABE7 (adenine base editor 7).
Como ya he dicho más arriba su trabajo ha descubierto que el editor de bases de citosina (BE3) genera unas 20 veces más variantes de un solo nucleótido no deseadas que el editor de bases de adenina (ABE7). La mayoría de las mutaciones no deseadas (off-target) son independientes de la guía de ARN, lo que implicaba que su posición no fue debida a un error de la dCas9, sino de la actividad aleatoria no deseada de la desaminasa fusionada a la dCas9.
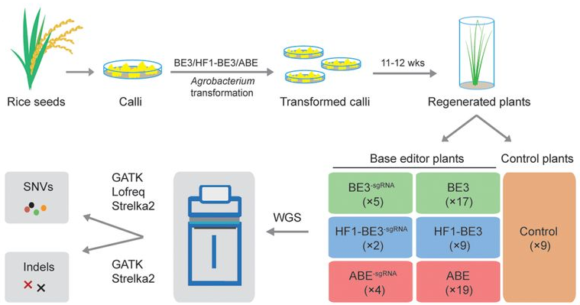
Y el tercer artículo en Science está liderado por Caixia Gao, Institute of Genetics and Developmental Biology of the Chinese Academy of Sciences, Beijing, cuyos primeros coautores son Shuai Jin, Yuan Zong y Qiang Gao. Han realizado una secuenciación de genoma completo para buscar mutaciones no deseadas al usar los editores de bases de la citosina (CBE), C>T, y de la adenina (ABE), A>G, en concreto BE3 (third-generation base editor), high-fidelity BE3 (HF1-BE3), y ABE. Se usaron 14 editores de bases para editar 11 sitios del genoma del arroz. También se usaron las técnicas de edición de bases sin ARN guía, así como dos grupos de plantas de control.

El número medio de variantes de un solo nucleótido de tipo C>T por planta fueron 203 (BE3), 347 (HF1-BE3), 88 (ABE) y 105 (control group). Por ello concluyen que los cambios C>T en BE3 y HF1-BE3 en plantas son 94.5% y 231.9% más frecuentes que en las plantas de control. Más aún, la mayoría de las variantes de un solo nucleótido de las técnicas no deseadas de tipo C>T de las técnicas BE3 y HF1-BE3 no aparecieron donde predijeron las técnicas in silico (Cas-OFFinder). Aparecieron distribuidos por todo el genoma. Por tanto, Gao y sus colegas concluyen que se debe optimizar las técnicas BE3 y HF1-BE3, o usar en su lugar las técnicas ABE.
Como nos cuentan Kempton y Qi en Science, los resultados de los artículos liderados por Yang y Gao no son del todo sorprendentes, dadas las propiedades de la desaminasas usadas. El sistema de editor de bases BE3 utiliza la proteína APOBEC de rata (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like, o enzima editora de la apolipoproteína B mediante ARNm similar a un polipéptido catalítico), una citidina desaminasa que puede unirse al ADN monocatenario independientemente de Cas9. Esto podría explicar las mutaciones aleatorias no deseadas observadas.
Por el contrario, el editor de bases de adenina utiliza como desaminasa una proteína bacteriana TadA modificada (tRNA-specific adenosine deaminase, o desaminasa de la adenosina específica del ARN de transferencia). A diferencia de APOBEC, TadA carece de la capacidad de unirse al ADN por sí sola y, por lo tanto, su actividad está más restringida a los sitios de unión especificados por la guía de ARN adjunta a Cas9. Los editores de bases de citosina son muy útiles, por lo que se requieren futuros estudios que mejoren su especificidad, limitando de alguna forma la actividad de la desaminasa fuera de la unión de Cas9. Sin lugar a dudas habrá avances en este sentido en los próximos años.
Esta tecnica serviría para curar
Sordera genetica???
Belén, en un futuro aún lejano. Recuerda, las aplicaciones clínicas de las técnicas CRISPR en humanos aún están muy lejanas. Por ahora solo se usan en investigación.
¿Se ha probado ésta técnica en SMA (spinal muscular atrophy)? Tengo dos hijos con un SNP en uno de los alelos que codifica para SMN1. Y claramente esta técnica es la cura para la enfermedad. Aguardo su respuesta. Muchas gracias
Gabriela, hasta donde me consta solo se ha usando a nivel preclínico. Lo siento, pero las aplicaciones clínicas de las técnicas CRISPR en humanos aún están muy lejanas. Por ahora solo se usan en investigación.
Esto significa que nunca podremos usar ninguna variante de criper casX sin que produzca off target?
No, Alicia, solo que queda mucha investigación por realizar para su uso en humanos. La técnica de edición CRISPR nació en 2012. Todavía es muy joven.